

Produtos que estão no dia a dia de todos, incluindo roupas, mesmo infantis, como carrinho de nenê, produtos anti-aderentes e muitos e muitos outros.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3365860
Endocr Rev . 2012 jun; 33 (3): 378–455.Publicado online 2012 14 de março. Doi: 10.1210 / er.2011-1050PMCID: PMC3365860PMID: 22419778
Laura N. Vandenberg , Theo Colborn , Tyrone B. Hayes , Jerrold J. Heindel , David R. Jacobs, Jr. , Duk-Hee Lee , Toshi Shioda , Ana M. Soto , Frederick S. vom Saal , Wade V. Welshons , R. Thomas Zoeller e John Peterson Myers
[NOTA DO WEBSITE: além de honrar Theo Colborn, autora do livro ‘O Futuro Roubado’, inspiradora deste site, em um de seus últimos trabalhos, de 2012, honramos este material porque também traz uma definitiva mudança de um paradigma toxicológico. Mostra como estas moléculas reconhecidas hoje como sendo disruptores endócrinos atuam em nossos corpos. Como elas são mimetizadoras de hormônios, é lógico que a dose não estará condicionada à ‘quantidade’ porque hormônios agem em mínimas porções. Assim, neste caso, não é a dose que determina se é ou não veneno -axioma da Idade Média de Paracelsus-, mas a simples presença destas moléculas já determinam sua interferência hormonal. Vale ressaltar que estas moléculas por isso, feminizam machos, geram enfermidades nas fêmeas, como tiroidiana e câncer de mama, além de outras. Este material é importante para todos nós, os consumidores, entendermos porque devemos imediatamente extirpar, de nossas vidas e do ambiente planetário, todas estas substâncias. Para ruptura de nossa atual visão da toxicologia, anexamos estes quadros abaixo, ficando totalmente – visível -, literalmente, a relação ‘quantidade’ e envenenamento.]
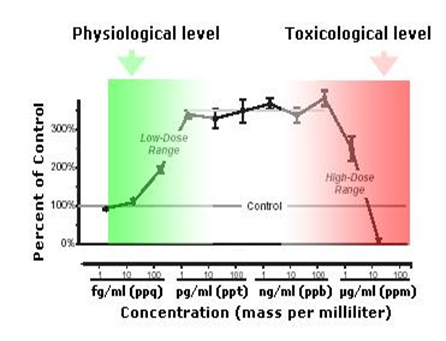

Resumo
Por décadas, estudos de produtos químicos disruptores endócrinos (Endocrine Disruptors Compounds/EDCs) desafiaram os conceitos tradicionais da toxicologia, em particular o dogma de que “a dose é que faz o veneno”, já que os EDCs podem ter efeitos em doses baixas, não previstas por efeitos em doses mais altas.
Aqui, revisamos dois conceitos principais nos estudos de EDC: baixa dose e curva não-monotônica (nt.: ver gráfico esquemático acima). Efeitos de baixa dose foram definidos pelo Programa Nacional de Toxicologia como aqueles que ocorrem na faixa de exposições humanas ou efeitos observados em doses abaixo daquelas usadas para estudos toxicológicos tradicionais.
Revisamos os dados mecanísticos para efeitos de doses baixas e usamos uma abordagem de peso de evidência para analisarmos cinco exemplos da literatura da EDC. Além disso, exploramos curvas de dose-resposta não-monotônicas (nt.: gráfico acima), definida como uma relação não linear entre dose e efeito, em que a inclinação da curva muda de sinal em algum lugar dentro da faixa de doses examinadas.
Fornecemos uma discussão detalhada dos mecanismos responsáveis pela geração destes fenômenos, além de centenas de exemplos da literatura sobre cultura de células, animais e epidemiologia. Ilustramos que respostas não monotônicas e efeitos de baixas doses são notavelmente comuns em estudos de hormônios naturais e EDCs. Se doses baixas de EDCs influenciam certos distúrbios humanos, não é mais conjectura porque estudos epidemiológicos mostram que as exposições ambientais aos EDCs estão associadas a doenças e deficiências humanas. Concluímos que, quando ocorrem curvas dose-resposta não monotônicas, os efeitos de doses baixas não podem ser previstos pelos efeitos observados em doses altas.
Portanto, mudanças fundamentais nos testes químicos e na determinação de segurança são necessárias para proteger a saúde humana.
- Introdução
- Antecedentes: exposição a baixas doses
- Antecedentes: NMDRCs/nonmonotonic dose-response curves (nt.: curvas de dose-resposta não-monotônicas)
- Estudos de baixa dose: uma década após a avaliação do painel NTP/National Toxicology Program (nt.: Programa Nacional de Toxicologia dos EUA)
- Por que examinar estudos de baixa dose agora?
- Mecanismos para efeitos de baixa dose
- Posição intrauterina e gêmeos humanos: exemplos de efeitos naturais de baixa dose
- Demonstrando efeitos de baixa dose usando uma abordagem WoE/weight of evidence (nt.: peso de evidência)
- Uso de uma abordagem WoE em estudos de baixa dose de EDC
- Refutando estudos de baixa dose: critérios necessários para aceitação de estudos que não encontram efeito
- BPA e próstata: efeitos contestados em doses baixas?
- BPA e glândula mamária: evidência indiscutível de efeitos de baixa dose
- Outro exemplo controverso de doses baixas: atrazina e desenvolvimento sexual de anfíbios
- Dioxina e espermatogênese: efeitos de doses baixas do disruptor endócrino mais potente?
- Perclorato e tireoide: efeitos de baixa dose em humanos?
- Resumo de baixa dose
- Não monotonicidade em estudos EDC
- Por que a não-monotonicidade é importante?
- Mecanismos para NMDRCs
- Exemplos de não monotonicidade
- Resumo do NMDRC
- Implicações dos efeitos de baixa dose e não monotonicidade
- Design experimental
- Ciência regulatória
- Saúde humana
- Animais selvagens
- Sumário
I. Introdução
Esta revisão se concentra em duas questões principais no estudo de produtos químicos disruptores endócrinos (EDCs): exposições a baixas doses e curvas de resposta à dose não monotônicas (NMDRCs). Esses conceitos são inter-relacionados, e as NMDRCs são especialmente problemáticas para avaliar os possíveis impactos da exposição quando a não-monotonicidade é evidente em níveis de exposição inferiores aos que são normalmente usados em avaliações toxicológicas. Para maior clareza da apresentação, examinaremos primeiro cada um dos conceitos separadamente.
A. Antecedentes: baixa dose de exposição
Está bem estabelecido na literatura endócrina que os hormônios naturais agem em concentrações séricas extremamente baixas, tipicamente na faixa de picomolar (nt.: são 11 zeros e doze casas, depois da vírgula = 0, 000000000001. Para comparar, um miligrama tem 5 zeros e seis casas = 0,000001) a nanomolar (nt.: são 8 zeros e 9 casas, depois da vírgula = 0.000000001). Muitos estudos publicados na literatura, revisada por pares, documentam que os EDCs podem atuar na faixa nanomolar a micromolar (nt.: são 5 zeros e seis casas, depois da vírgula = 0, 000001), e alguns mostram atividade nos níveis picomolar.
1. O que se entende por dose baixa?
Em 2001, a pedido da Agência de Proteção Ambiental dos EUA (EPA/Environmental Protection Agency), o Programa Nacional de Toxicologia (NTP/NAtional Toxicology Program) reuniu um grupo de cientistas para realizar uma revisão da literatura sobre EDC em baixa dose ( 1 ). Naquele momento, o painel NTP definiu efeitos de baixa dose como qualquer mudança biológica: 1) ocorrendo na faixa de exposições humanas típicas; ou 2) ocorrendo em doses inferiores às normalmente usadas em protocolos de teste padrão, ou seja , doses abaixo das avaliações testadas na toxicologia tradicional ( 2 ). Outras definições de dose baixa incluem: 3) uma dose abaixo da dose mais baixa na qual uma alteração biológica (ou dano) para um produto químico específico foi medida no passado, ou seja ,qualquer dose abaixo do nível mais baixo de efeito observado ou do nível mais baixo de efeito adverso observado (LOAEL/lowest observed adverse effect level) ( 3 ); ou 4) uma dose administrada a um animal que produz concentrações sanguíneas desse produto químico na faixa do que foi medido na população humana em geral ( isto é, não exposto ocupacionalmente, e frequentemente referido como uma dose ambientalmente relevante porque cria uma dose interna relevante para as concentrações do produto químico medido em humanos) ( 4 , 5 ). Esta última definição leva em conta as diferenças no metabolismo química e farmacocinética (isto é, absorção, distribuição e excreção do produto químico) entre espécies e reduz a importância da via de exposição, comparando diretamente concentrações similares de sangue ou outros tecidos nos sistemas modelo e paradigmas experimentais. Embora essas definições diferentes possam parecer bastante semelhantes, o uso de apenas um único produto químico bem estudado, como o bisfenol A (BPA), mostra como essas definições produzem pontos de corte diferentes para concentrações de exposição consideradas baixas doses (tabela 1). Para muitos produtos químicos, incluindo EDCs, um grande número de estudos atende aos critérios para estudos de doses baixas, independentemente de o ponto de corte para uma dose baixa ter sido baseado na faixa de exposições humanas típicas, doses usadas na toxicologia tradicional ou doses que usam uma medida interna da carga corporal.
Tabela 1.
Definições de baixas doses e doses de corte: BPA e DEHP como exemplos
| Químico | Intervalo estimado de exposições humanas | Doses abaixo do NOAEL | Doses abaixo do LOAEL | Doses administradas (a animais) que produzem níveis sanguíneos em humanos típicos |
|---|---|---|---|---|
| BPA | 0,4–5 μg / kg · d ( 679 ) | Nenhum NOAEL foi estabelecido em estudos toxicológicos ( 38 ) | <50 mg / kg · d ( 38 ) | ∼400 μg / kg · d em roedores e primatas não humanos ( 4 , 253 ) |
| DEHP | 0,5–25 μg / kg · d ( 680 ) | <5,8 mg / kg · d ( 681 , 682 ) | <29 mg / kg · d ( 681 , 682 ) | Desconhecido |
Se doses baixas de EDCs influenciam doença, e agora passam a ser uma questão que se estende muito além da bancada do laboratório é porque também estudos epidemiológicos mostram que as exposições ambientais a estes produtos químicos estão sendo associadas a distúrbios em seres humanos (ver, por exemplos, Refs. 6 – 16 ). Embora associações de doenças tenham sido historicamente observadas em indivíduos expostos a grandes concentrações de EDCs, após acidentes industriais ( 17 – 19 ) ou via aplicações ocupacionais ( 20 – 22), estudos epidemiológicos recentes revelam ligações entre baixas concentrações ambientalmente relevantes e a prevalência de doenças. Com os extensos estudos de biomonitoramento realizados pelos Centros dos EUA para Controle e Prevenção de Doenças (CDC/Centers for Disease Control and Prevention) ( 23 , 24 ) e pesquisas ambientais semelhantes realizadas na Europa ( 25 ) e em outros lugares ( www.statcan.gc.ca/concepts/hs-es/measure-mesures-eng.htm ), o conhecimento sobre exposições ambientais a EDCs e suas associações com distúrbios da saúde humana, aumentou substancialmente.
Os efeitos de baixa dose receberam atenção considerável das comunidades científica e reguladora, especialmente quando examinados para substâncias químicas únicas e bem estudadas, como o BPA ( 4 , 27 – 32 ). A literatura de baixas doses como um todo, no entanto, não foi cuidadosamente examinada por mais de uma década (nt.: esta afirmativa é em razão de que o livro da Dra. Theo Colborn e outros, ‘Our Stolen Future‘ / ‘O Futuro Roubado’, lançado em 1994, já era uma compilação de estudos que vinham desde os anos 80, demonstrando os efeitos destas moléculas em baixas doses). Além disso, esse corpo de literatura foi desconsiderado ou considerado insignificante por muitos ( 33 , 34 ). Desde a revisão do NTP da literatura sobre baixas doses em 2001 ( 2), um grande número de dados foi publicado, incluindo: 1) exemplos impressionantes adicionais de efeitos de baixa dose de exposições a EDCs bem caracterizados, bem como outros produtos químicos; 2) uma compreensão dos mecanismos responsáveis por esses efeitos de baixa dose; 3) exploração da não monotonicidade em sistemas in vivo e in vitro; e 4) suporte epidemiológico para efeitos de baixa dose e NMDRCs/nonmonotonic dose-response curves.
2. O termo dose baixa é um nome impróprio?
Hormônios endógenos são ativos em doses muito baixas, dentro e abaixo da variação de picomolar para estrogênios endógenos e drogas estrogênicas, ao passo que estes disruptores endócrinos ambientais que mimetizam estrogênios são geralmente ativos nos níveis de nanomolar e micromolar (para exemplos, ver, Refs. 35 – 38 ). Mesmo que alguns mostrem efeitos em concentrações ainda mais baixas ( 39 – 41). É importante ressaltar que as definições acima não levam em consideração a potência ou eficácia do produto químico em questão, um tópico que será discutido em mais detalhes abaixo. Em vez disso, a dose baixa fornece uma definição operacional, na qual as doses que estão na faixa de exposição humana ou doses abaixo daquelas tradicionalmente testadas em estudos toxicológicos, são consideradas baixas. Para ficar claro, nenhuma destas definições sugere que uma única concentração possa ser definida como um ponto de corte de baixa dose para todos os produtos químicos. Usando as definições acima, para alguns produtos químicos, doses baixas podem estar na faixa de nanogramas por quilograma. Mas para a maioria dos produtos químicos, doses na faixa tradicional de micro e miligramas por quilograma, podem ser consideradas doses baixas nas abordagens tradicionais para testarem normalmente os produtos químicos já que não examinam doses abaixo do intervalo de doses em miligramas por quilograma.
[NOTA DA WEBSITE: por ser esta publicação científica bem abrangente, completa, profunda, ampla e por isso complexa para o cidadão comum, resolvemos traduzir a parte inicial até este ponto e agora interrompemos, mas agregando a parte final, mais conclusiva, do artigo para melhor compreensão. Após esta inclusão passará a estar a totalidade do texto original em inglês para que os leitores que quiserem aprofundar seu conhecimento, possam desfrutar de seus conteúdos. Para quem não sabe inglês, mas quer ler todo o artigo em português, reconecta o link original e quando a janela acima, à direita, aparecer perguntando se quer traduzir para o português, clique concordando. Ressaltamos que é uma tradução mecânica e por isso pode incorrer em erros de compreensão. Mesmo assim, para quem desconhece tudo sobre todo o tema, já dará, para o leigo, uma visão mais ampliada da matéria.]
………………………………………………..
………………………………………………..
V. Resumo
Em conclusão, fornecemos centenas de exemplos que mostram claramente que NMDRCs e efeitos de baixas doses são comuns em estudos de hormônios e EDCs. Examinamos cada uma dessas questões separadamente e fornecemos explicações mecanicistas e exemplos de ambas. Esses tópicos estão relacionados, mas devem ser examinados individualmente para serem entendidos. O conceito de não-monotonicidade é essencial para o campo da ciência em saúde ambiental, porque quando as NMDRCs ocorrem, os efeitos de baixas doses não podem ser previstos pelos efeitos observados em altas doses. Além disso, a descoberta de que produtos químicos têm efeitos adversos em animais e humanos na faixa de exposições ambientais, indica claramente que doses baixas não podem ser ignoradas.
Para encerrar, incentivamos cientistas e editores de periódicos a publicarem dados demonstrando as NMDRCs e efeitos de baixas doses, mesmo que o mecanismo exato de ação ainda não tenha sido elucidado. Isso é importante porque o estudo dos EDCs é uma especialidade crescente que atravessa muitos campos científicos e os cientistas que trabalham ou regulam os EDCs devem apreciar e reconhecer a existência de NMDRCs e efeitos de baixa dose e terem acesso a essas importantes informações. Recomendamos ainda testes e vigilância de segurança amplamente expandidos e generalizados para detectarem possíveis efeitos adversos desta ampla classe de produtos químicos (nt.: muitíssimos deles usados especificamente para o consumo de alimentos humanos e de contato permanente com mulheres grávidas e crianças). Antes do desenvolvimento de novos produtos químicos, uma ampla gama de doses, estendendo-se para a faixa de baixas doses, devem ser totalmente testadas. E, finalmente, prevemos que os conceitos e resultados empíricos que apresentamos neste artigo levarão a muito mais colaborações entre cientistas pesquisadores em laboratórios acadêmicos e governamentais de todo o mundo, que surgirão projetos de estudos cada vez mais sofisticados bem como o que produzimos aqui facilitará aqueles que tomam decisões regulatórias. E que possam levar, por fim, pelas ações tomadas à luz destas informações, à diminuição do uso de EDCs e, finalmente, que os impactos na saúde das pessoas e da vida selvagem serão evitados.
……………………………………………….
……………………………………………….
B. Background: NMDRCs
We have defined low-dose studies according to the definitions established by the NTP panel of experts (2). However, because the types of endpoints that are typically examined at high doses in toxicological studies are often different from the types of endpoints examined in low-dose studies, one cannot assume that an effect reported in the low-dose range is necessarily different from what would be observed at higher doses. For example, low doses of a chemical could affect expression of a hormone receptor in the hypothalamus, an endpoint not examined in high-dose toxicology testing, and high doses could similarly affect this same endpoint (but are likely to be unreported because high doses are rarely tested for these types of endpoints). Thus, the presence of low-dose effects makes no assumptions about what has been observed at higher concentrations. (As discussed elsewhere, for the majority of chemicals in commerce, there are no data on health effects and thus no established high- or low-dose range.) Therefore, low-dose effects could be observed at the lower end of a monotonic or linear dose-response curve.
In contrast, the definition of a NMDRC is based upon the mathematical definition of nonmonotonicity: that the slope of the dose-response curve changes sign from positive to negative or vice versa at some point along the range of doses examined (42). Often NMDRCs have a U- or inverted U-shape (43); these NMDRCs are thus also often referred to as biphasic dose-response curves because responses show ascending and descending phases in relation to dose. Complex, multiphasic curves have also been observed (41, 44, 45). NMDRCs need not span from true low doses to high (pharmacologically relevant) doses, although experiments with such a broad dose range have been performed for several EDCs; the observation of nonmonotonicity makes no assumptions about the range of doses tested. Examples of NMDRCs from in vitro cell culture and in vivo animal experiments, as well as epidemiological examples, are presented in detail later in this review (see Sections III.C.1–3). Additional examples of NMDRCs are available in studies examining the effects of vitamins and other essential elements on various endpoints (see for example (46); these will not be examined in detail in this review due to space constraints.
NMDRCs present an important challenge to traditional approaches in regulatory toxicology, which assume that the dose-response curve is monotonic. For all monotonic responses, the observed effects may be linear or nonlinear, but the slope does not change sign. This assumption justifies using high-dose testing as the standard for assessing chemical safety. When it is violated, high-dose testing regimes cannot be used to assess the safety of low doses.
It should be noted that both low dose and nonmonotonicity are distinguished from the concept of hormesis, which is defined as a specific type of response whereby “the various points along [the dose response] curve can be interpreted as beneficial or detrimental, depending on the biological or ecological context in which they occur” (47). Estimations of beneficial or adverse effects cannot be ascertained from the direction of the slope of a dose-response curve (48–50). In their 2001 Low Dose Peer Review, the NTP expert panel declined to consider whether any effect was adverse because “in many cases, the long-term health consequences of altered endocrine function during development have not been fully characterized” (2). There are still debates over how to define adverse effects (51–53), so for the purposes of this review, we consider any biological change to be an effect. Importantly, most epidemiological studies are by definition examining low doses (unless they are focusing on occupationally exposed individuals), and these studies typically focus on endpoints that are accepted to be adverse for human health, although some important exceptions exist (54–56).
Finally, it is worth noting that any biological effect, whether it is observed to follow linear relationships with administered dose or not, provides conclusive evidence that an EDC has biological activity. Thus, other biological effects are likely to be present but may remain undetected or unexamined. Many EDCs, including those used as pesticides, were designed to have biological effects (for example, insecticides designed to mimic molting hormone). Thus, the question of whether these chemicals have biological effects is answered unequivocally in their design; the question is what other effects are induced by these biologically active agents, not whether they exist.
C. Low-dose studies: a decade after the NTP panel’s assessment
In 2000, the EPA requested that the NTP assemble a panel of experts to evaluate the scientific evidence for low-dose effects and dose-response relationships in the field of endocrine disruption. The EPA proposed that an independent and open peer review of the available evidence would allow for a sound foundation on which the EPA could “determine what aspects, if any, of its standard guidelines for reproductive and developmental toxicity testing [would] need to be modified to detect and characterize low-dose effects” (2). The NTP panel verified that low-dose effects were observed for a multitude of endpoints for specific EDCs including diethylstilbestrol (DES), genistein, methoxychlor, and nonylphenol. The panel identified uncertainties around low-dose effects after exposure to BPA; although BPA had low-dose effects on some endpoints in some laboratories, others were not found to be consistent, leading the panel to conclude that it was “not persuaded that a low-dose effect of BPA has been conclusively established as a general or reproducible finding” (2).
Since the NTP’s review of low-dose endocrine disruptor studies, only a few published analyses have reexamined the low-dose hypothesis from a broad perspective. In 2002, R. J. Witorsch (57) analyzed low doses of xenoestrogens and their relevance to human health, considering the different physiologies associated with pregnancy in the mouse and human. He proposed that low doses of endocrine disruptors would not likely affect humans because, although low-dose effects had been observed in rodents, the hormonal milieu, organs controlling hormonal release, and blood levels of estrogen achieved are quite different in humans. There are, of course, differences in hormones and hormone targets between rodents and humans (58), but the view that these differences negate all knowledge gained from animal studies is not supported by evolutionary theory (59–61). This human-centered stance argues against the use of animals for any regulatory testing (62) and runs counter to the similarities in effects of EDCs on humans and animals; rodents proved to be highly predictive of the effects of DES on humans (63, 64). In a striking example, studies from mice and rats predicted that gestational exposure to DES would increase mammary cancer incidence decades before women exposed in utero reached the age where this increase in risk was actually observed (65–67).
In 2007, M. A. Kamrin (68) examined the low-dose literature, focusing on BPA as a test case. He suggested that three criteria were required to support the low-dose hypothesis. First is reproducibility, which he defined as “the same results are seen from the same causes each time a study is conducted.” Furthermore, he proposed that the dose response for the effects must be the same from study to study. Second is consistency, which he defined as the results all fitting into a pattern, whereby the results collected from multiple species and under variable conditions all show the same effect. And third is proper conduct of studies, which he defined as including the appropriate controls and performance under suitable experimental conditions as well as the inclusion of multiple doses such that a dose-response curve can be obtained.
Although we and others (69–72) agree with the use of these criteria (reproducibility, consistency, and proper experimental design), there are significant weaknesses in the logic Kamrin employed to define these factors. First, suggesting that reproducibility is equivalent to the same results obtained each time a study is conducted is unrealistic and not a true representation of what is required of replication. As has been discussed in other fields, “there is no end to the ways in which any two experiments can be counted as the same — or different … All experiments are the same in respect of their being experiments; they are all different by virtue of being done at different places, at different times, by different people, with different strains of rat, training regime, and so on” (73).
Furthermore, according to the Bradford-Hill criteria, a set of requirements accepted in the field of epidemiology to provide adequate evidence of a causal relationship between two factors, a single negative result (or even several studies showing negative results) cannot negate other studies that show adverse effects (74). Essentially, all scientists know that it is very easy for an experiment to find no significant effects due to a myriad of reasons; it is more difficult to actually find effects, particularly when using highly sophisticated techniques (69).
Second, the concept of consistency as a pattern that can be derived from all results is one we will use below, using a weight-of-evidence (WoE) approach and several specific examples. However, Kamrin’s proposed idea that every study must show the same effect has the same weaknesses as discussed for the proposed definition of reproducibility and does not acknowledge the obvious differences in many species and strains. It also suggests that the identification of a single insensitive strain could negate any number of positive studies conducted with appropriate animal models (75).
And finally, Kamrin suggested that only studies with appropriate controls should be used for analyses, a criterion we agree should be followed. However, his own scrutiny of the low-dose animal literature fails to do so (68). He also suggested that studies use multiple doses so that a dose-response curve can be obtained. Although studies using a single dose can be informative, we agree that dose-response relationships provide important information to researchers and risk assessors alike. However, this requirement is not helpful if there is an insistence on observing a linear response; as we discuss in depth in this review, there are hundreds of examples of nonmonotonic and other nonlinear relationships between dose and endpoint. These should not be ignored.
In 2004, Hayes (76) reviewed the available literature concerning the effects of atrazine on amphibian development, with a specific focus on the effect of ecologically relevant doses of this EDC on malformations of the gonads and other sexually dimorphic structures; in the case of aquatic exposures, it can be difficult to determine what a cutoff for a low dose would be; thus, Hayes focused on studies examining the effects of atrazine at levels that had been measured in the environment. He reviewed the results produced by several labs, in which it was independently demonstrated that low concentrations of atrazine produced gonadal abnormalities including hermaphroditism, males with extra testes, discontinuous gonads, and other defects. Hayes’ work also clearly addressed the so-called irreproducibility of these findings by analyzing the studies that were unable to find effects of the pesticide; he noted that the negative studies had multiple experimental flaws, including contamination of the controls with atrazine, overcrowding (and therefore underdosing) of experimental animals, and other problems with animal husbandry that led to mortality rates above 80%.
In 2006, vom Saal and Welshons (77) examined the low-dose BPA literature, identifying more than 100 studies published as of July 2005 that reported significant effects of BPA below the established LOAEL, of which 40 studies reported adverse effects below the 50 μg/kg · d safe dose set by the EPA and U.S. Food and Drug Administration (FDA); all of these studies would be considered low dose according to the NTP’s definition (2). The authors proposed that these examples should be used as evidence to support the low-dose hypothesis. Furthermore, this publication detailed the similarities among the studies that were unable to detect any effects of low doses of BPA and established a set of criteria required to accept negative studies. We have adapted the criteria detailed by Hayes (76) and vom Saal and Welshons (77) to produce a set of requirements for low-dose studies; these criteria are described in some detail below.
D. Why examine low-dose studies now?
The developmental origins of health and disease hypothesis originated from studies showing that fetal DES exposure could cause severe malformations and cancers of the reproductive tract, and other studies demonstrating that fetal malnutrition could lead to adult diseases including metabolic syndrome, diabetes, and increased stroke incidence (78–81). Since that time, the developmental origins of health and disease hypothesis has been extended to address whether diseases that are increasing in prevalence in human populations could be caused by developmental exposures to EDCs (67, 82–85). Evidence from the animal literature has been tremendously informative about the effects of EDC exposures early in development and has driven new hypotheses to be tested in epidemiology studies (86). Studies including several discussed in this review provide supportive evidence that the fetal and neonatal periods are specifically sensitive to chemicals that alter endocrine signaling and that EDCs could be contributing to a range of diseases.
Strong, reliable, and reproducible evidence documents the presence of low concentrations of EDCs and other chemicals in human tissues and fluids, as well as in environmental samples (28, 87–89). These studies indicate that samples collected from humans and the environment typically contain hundreds of contaminants, usually in the parts-per-billion (ppb) range (90, 91). The obvious question with potentially large public health implications is whether these concentrations are so low as to be irrelevant to human health. The fact that epidemiological analyses (reviewed in Section III.C.3) repeatedly find associations between the measured concentrations in human samples and disease endpoints suggests it is inappropriate to assume the exposures are too low to matter. That is especially the case given the empirical data (reviewed in Section II.A) from animal and cell culture experiments showing effects can be caused by concentrations comparable (and sometimes below) what is measured in humans and also the detection of NMDRCs in some of those same experiments.
In the human biomonitoring field, large databases such as the CDC’s National Health and Nutrition Examination Survey (NHANES) have allowed researchers to make comparisons between groups of individuals with various exposure criteria; some of these studies will be addressed in detail in subsequent sections of this review. Although by definition these databases examine low-dose exposures, their use has been the subject of significant debate. Because of the large number of chemicals that have been measured (>300 in the most recent NHANES by the CDC) and the large number of health outcomes and other disease-related data collected from the individuals that donated biological samples, it has been argued that the number of possible associations that could be made would lead to a significant number of false positives (92); thus, associations could be found simply because of extensive data dredging. This has led some to suggest that these studies as a whole should be rejected (93, 94).
In response to these criticisms, epidemiologist Jan Vandenbroucke (95) notes, “researchers do not mindlessly grind out one analysis after another”; the examination of these databases for associations between chemical exposures and health effects does not entail the statistical comparison between all possible factors, calculated as some 8800 comparisons in the CDC’s NHANES database (92). Instead, epidemiologists typically focus on a select number of comparisons that address relationships between chemicals and diseases identified a priori (96, 97), often because of mechanistic data obtained in laboratory animals or in vitro work with human and animal cells and tissues. Repeated findings of links between EDC exposures and diseases in epidemiological analyses of biomonitoring data based on a priori hypotheses suggests these relationships should not be rejected as a statistical artifact and, instead, should be the basis for significant concern that low-dose effects can be detected in the general population (85, 98).
E. Mechanisms for low-dose effects
The endocrine system is particularly tuned to respond to very low concentrations of hormone, which allows an enormous number of hormonally active molecules to coexist in circulation (38). As a ligand-receptor system, hormones act by binding to receptors in the cell membrane, cytosol, or the nucleus. The classical effects of nuclear hormone receptors influence gene expression directly, although rapid nongenomic actions at membrane-associated receptors are now well documented and accepted. Membrane receptors are linked to different proteins in the cell, and binding to these receptors typically changes cellular responses in a rapid fashion (99), although the consequence of a rapid signaling event could be the activation of a nuclear transcription factor, leading to responses that take longer to detect. Peptide hormones can also influence gene expression directly (see Refs. 100 and 101 for examples).
There are several means by which the endocrine system displays specificity of responses to natural hormones. Many hormone receptors are expressed specifically in a single or a few cell types (for example, receptors for TSH are localized to the thyroid), whereas some (like thyroid hormone receptors) are found throughout the body (102). For receptors that are found in multiple cell types, different effects are produced in part due to the presence of different coregulators that influence behaviors of the target genes (103–105). And finally, some hormones have multiple receptors [for example estrogen receptor (ER)α and ERβ], which are expressed in different quantities in different cell types and organs and can produce variable effects on gene expression or cellular phenomena (cell proliferation vs. apoptosis) (102, 106).
The typical physiological levels of the endogenous hormones are extremely low, in the range of 10–900 pg/ml for estradiol, 300–10,000 pg/ml for testosterone, and 8–27 pg/ml for T4 (see Table 2). Importantly, steroid hormones in the blood are distributed into three phases: free, representing the unconjugated, unbound form; bioavailable, representing hormones bound to low-affinity carrier proteins such as albumin; and inactive, representing the form that is bound to high-affinity binding proteins such as SHBG or α-fetoprotein (38) (Fig. 1A). When the circulating levels in blood are corrected for the low fraction of the hormones that are not bound to serum binding proteins, the free concentrations that actually bring about effects in cells are even lower, for example 0.1–9 pg/ml for estradiol. Concentrations of active hormones will vary based on the age and physiological status of the individual (i.e. plasma testosterone levels are less than 1 ng/ml in male children but increase to approximately 5–7 ng/ml in adulthood; during menses, estradiol levels are typically less than 100 pg/ml, but just before ovulation, they spike to 800 pg/ml; etc.) (107, 108). Of course, it should be noted that active concentrations of natural hormones vary somewhat from species to species and can even vary between strains of the same species (109).
Table 2.
Ranges of endogenous hormones in humans (from Ref. 108)
| Hormone | Free concentration (females) | Total concentration (females) | Free concentration (males) | Total concentration (males) |
|---|---|---|---|---|
| Cortisol | 20–300 ng/ml | 20–300 ng/ml | ||
| Estradiol | 0.5–9 pg/ml (adult female) | <20 pg/ml (prepubertal) | 10–60 pg/ml (adult) | |
| 20–800 pg/ml (premenopausal) | ||||
| <30 pg/ml (postmenopausal) | ||||
| Progesterone | 0.2–0.55 ng/ml (prepubertal) | 0.1–0.4 ng/ml (prepubertal) | ||
| 0.02–0.80 ng/ml (follicular phase) | 0.2–2 ng/ml (adult) | |||
| 0.90–4 ng/ml (luteal phase) | ||||
| <0.5 ng/ml (postmenopausal) | ||||
| Insulin | 0–250 pmol/liter | 0–250 pmol/liter | ||
| GH | 2–6 ng/ml | 2–6 ng/ml | ||
| Prolactin | 0–15 ng/ml | 0–10 ng/ml | ||
| Testosterone | 9–150 pg/ml (adult) | 0.3–250 ng/ml | ||
| Thyroid hormone | 8–30 pg/ml (10–35 pm) | 8–30 pg/ml (10–35 pm) | ||
| TSH | 0.5–5 μU/ml | 0.5–5 μU/ml |

Characteristics and activities of natural hormones. A, This schematic depicts a typical relationship of three phases of circulating hormones: free (the active form of the hormone), bioavailable (bound weakly to proteins such as albumin), and inactive (bound with high affinity to proteins such as SHBG). These three phases act as a buffering system, allowing hormone to be accessible in the blood, but preventing large doses of physiologically active hormone from circulating. With EDCs, there may be little or no portion maintained in the inactive phase. Thus, the entirety or majority of a circulating EDC can be physiologically active; the natural buffering system is not present, and even a low concentration of an EDC can disrupt the natural balance of endogenous hormones in circulation. B, Schematic example of the relationship between receptor occupancy and hormone concentration. In this theoretical example, at low concentrations, an increase in hormone concentration of x (from 0 to 1x) causes an increase in receptor occupancy of approximately 50% (from 0 to 50%, see yellow box.) Yet the same increase in hormone concentration at higher doses (from 4x to 5x) causes an increase in receptor occupancy of only approximately 4% (from 78 to 82%, see red box).
There are several reasons why endogenous hormones are able to act at such low circulating concentrations: 1) the receptors specific for the hormone have such high affinity that they can bind sufficient molecules of the hormone to trigger a response, 2) there is a nonlinear relationship between hormone concentration and the number of bound receptors, and 3) there is also a nonlinear relationship between the number of bound receptors and the strongest observable biological effect. Welshons and colleagues (38) describe how hormone concentration influences receptor occupancy: “receptor occupancy is never determined to be linear in relation to hormone concentration … At concentrations above the Kd [the dissociation constant for receptor-ligand binding kinetics], saturation of the response occurs first, and then at higher concentrations, saturation of receptors is observed.” What this means is that at low doses of hormone, a 10-fold increase in hormone concentration can have a 9-fold increase in receptor occupancy, whereas at high doses of hormone, a 10-fold increase in hormone concentration produces a less than 1.1-fold increase in receptor occupancy (38) (Fig. 1B). Thus, even moderate changes in hormone concentration in the low-dose range can produce substantial changes in receptor occupancy and therefore generate significant changes in biological effects. Welshons et al. (38) also note that a near-maximum biological response can be observed without a high rate of receptor occupancy, a situation that was previously termed the spare receptor hypothesis (110, 111); that is, the response mechanism saturates before all of the receptors are saturated. The presence of spare receptors is the basis for saying that these receptor systems are tuned to detect low concentrations that lead to occupancy of 0.1–10% of total receptors. Within this range of low receptor occupancy, there is high proportionality between changes in the free hormone concentration and changes in receptor occupancy, and a change in receptor occupancy by a ligand for the receptor is required to initiate changes in receptor-mediated responses (38).
There are additional reasons why natural hormones are active at low doses: 4) hormones have a strong affinity for their receptors (relative to affinity for other receptors) because many hormones are secreted from a single gland or site in the body but must have effects throughout the body in multiple tissues and 5) blood concentrations of hormones are normally pulsatile in nature, with the release of one hormone often controlled by the pulsatile release of another hormone (112, 113), and both the frequency and the amplitude of pulses modulate the biological response; hormones are also influenced by circadian rhythms, with dramatic differences in hormone secretion depending on the time of day (114, 115).
For many years, the mechanisms by which some environmental chemicals acted at low doses were not well understood. In 1995, the National Research Council appointed the Committee on Hormonally Active Agents in the Environment to address public concerns about the potential for adverse effects of EDCs on human health (116). At the time, work on understanding the mechanisms by which EDCs exert their effects was in its infancy, and in the executive summary, the committee stated, “Lack of knowledge about a mechanism does not mean that a reported effect is unconfirmed or unimportant, nor does demonstration of a mechanism document that the resulting effects are unique to that mechanism or are pervasive in natural systems.” Since that time, a tremendous amount of work has been dedicated to understanding the molecular mechanisms of action of EDCs, and in particular the mechanisms responsible for low-dose effects.
1. General mechanisms for EDC action
As discussed above, the endocrine system evolved to function when unbound physiologically active ligands (hormones) are present at extremely low doses (117). Because of shared receptor-mediated mechanisms, EDCs that mimic natural hormones have been proposed to follow the same rules and therefore have biological effects at low doses (38, 118). Similarly, EDCs that influence in any way the production, metabolism, uptake, or release of hormones also have effects at low doses, because even small changes in hormone concentration can have biologically important consequences (38, 119).
The estrogen-response mechanisms have been extensively studied with regard to the effects of endogenous estrogens and estrogenic drugs. In classical, genomic estrogen action, when endogenous estrogens bind to ER, those receptors bind to estrogen response element sequences or to a number of other response element sites adjacent to the genes directly responsive to estrogens; this binding influences transcription of estrogen-sensitive genes (120). Xenoestrogens produce the same reactions; these chemicals bind to ERs, which then initiate a cascade of molecular effects that ultimately modify gene expression. Therefore, for the actions of estrogenic EDCs, molecular mechanisms and targets are already known in some detail. Similar mechanisms are induced by the binding of androgens to the androgen receptor, or thyroid hormone agonists to the thyroid hormone receptor, among others. Additionally, there are EDCs that act as antagonists of these hormone systems, binding to a receptor, but not activating the receptor’s typical response, and preventing the binding or activity of the endogenous ligand. Finally, many EDCs bind to the receptor and trigger a response that is not necessarily the same as that triggered by the endogenous estrogens; these are termed selective ER modulators (SERMs). Ultimately, all of these actions occur at the level of the receptor.
Many studies have been dedicated to the understanding of which EDCs bind to which nuclear hormone receptors and how the binding affinities compare to the natural steroid. Thus, many of these chemicals have been classified as weak hormones. Yet studies have shown that, for example, the so-called weak estrogens like BPA can be equally potent as endogenous hormones in some systems, causing biological effects at picomolar levels (30, 38, 41, 121). Both endogenous estrogens and EDCs can bind to ER associated with the cell membrane [membrane-associated ER (mER)α and mERβ] that are identical to the nuclear ER (122–124), and a transmembrane ER called G-protein coupled receptor 30 that is structurally dissimilar to the nuclear ER and encoded by a distinct gene (125, 126). In many cells, 5–10% of total ERα and ERβ are localized to the plasma membrane (124); these membrane-associated receptors are capable of nongenomic steroid action in various cell types (30, 121, 127); thus, rapid and potent effects are well documented for many EDCs including BPA, DES, endosulfan, dichlorodiphenyldichloroethylene (DDE), dieldrin, and nonylphenol, among others (41, 128–130).
Finally, EDCs have other effects that are not dependent on binding to either classical or membrane-bound steroid hormone receptors. EDCs can influence the metabolism of natural hormones, thus producing differences in the amount of hormone that is available for binding either because more (or less) hormone is produced than in a typical system or because the hormone is degraded faster (or slower) than is normal. Other EDCs influence transport of hormone, which can also change the amount of hormone that is available for receptor binding. And EDCs can also have effects that are independent from known endocrine actions. One example is the effect of endogenous hormones and EDCs on ion channel activity. BPA, dichlorodiphenyltrichloroethane (DDT), DES, nonylphenol, and octylphenol have all been shown to disrupt Ca2+ channel activity and/or Ca2+ signaling in some cell types (131–134). This example illustrates how both natural hormones and EDCs can have hormonal activity via binding to nuclear hormone receptors but may also have unexpected effects via receptor-mediated actions outside of the classical endocrine system.
2. Mechanisms of EDC-induced low-dose actions
The various mechanisms by which EDCs act in vitro and in vivo provide evidence to explain how these chemicals induce effects that range from altered cellular function, to abnormal organ development, to atypical behaviors. Just as natural hormones display nonlinear relationships between hormone concentration and the number of bound receptors, as well as between the number of bound receptors and the maximal observable biological effect, EDCs obey these rules of binding kinetics (38). Thus, in a way, EDCs exploit the highly sensitive endocrine system and produce significant effects at relatively low doses.
To gain insight into the effects of natural hormones and EDCs on gene expression profiles, it is possible to calculate doses that produce the same effect on proliferation of cultured cells, i.e. the quantitative cellular response doses, and determine the effect of those doses on transcriptomal signature profiles. When this is done for estradiol and EDCs with estrogenic properties, the affected estrogen-sensitive genes are clearly different (135). However, an interesting pattern emerges: comparing profiles among only the phytoestrogens shows striking similarities in the genes up- and down-regulated by these compounds; profile comparisons between only the plastic-based estrogens also show similarities within this group. Yet even more remarkable is what occurs when the doses are selected not based on cell proliferation assays but instead on the ability of estradiol and estrogen-mimics to induce a single estrogen-sensitive marker gene. When doses were standardized based on marker gene expression, the transcriptomal signature profiles were very similar between estradiol and estrogen mimics (135). Taken together, these results suggest that the outcomes of these experiments are contextual to the normalization parameter and that marker gene expression and cell proliferation are not superimposable. This indicates that the biological level at which the effects of chemicals are examined (i.e. gene expression, cellular, tissue, organ, or organismal) can greatly impact whether low-dose effects are observed and how these effects are interpreted.
There are several other mechanisms by which low-dose activities have been proposed. One such possibility is that low doses of EDCs can influence the response of individuals or organs/systems within the body to natural hormones; thus, the exposed individual has an increased sensitivity to small changes in endogenous steroids, similar to the effects of intrauterine position (see Ref. 136 and Section I.F). In fact, several studies have shown that exposure to EDCs such as BPA during perinatal development can influence the response of the mammary gland to estrogen (137, 138) and the prostate to an estrogen-testosterone mixture similar to the concentrations produced in aging men (139–142). There is also evidence that EDCs work additively or even synergistically with other chemicals and natural hormones in the body (143–145). Thus, it is plausible that some of the low-dose effects of an EDC are actually effects of that exogenous chemical plus the effects of endogenous hormone.
Finally, it should be noted that during early development, the rodent fetus is largely, but not completely (146), protected from estrogen via the binding activity of α-fetoprotein, a plasma protein produced in high levels by the fetal liver (147). Some estrogen-like EDCs, however, bind very weakly to α-fetoprotein, and therefore, it is likely that this protein does not provide protection to the fetus during these sensitive developmental periods (36, 148). Furthermore, because EDCs may not bind to α-fetoprotein or other high-affinity proteins in the blood (148–150) and can have a higher binding affinity to proteins like albumin (compared with natural estrogens) (36, 149), the balanced buffer system in place for endogenous hormones may be disturbed (Fig. 1A). Thus, whereas only a portion of endogenous hormones are bioavailable, the entirety of a circulating EDC could be physiologically active.
The effects of hormones and EDCs are dependent on dose, and importantly, low (physiological) doses can be more effective at altering some endpoints compared with high (toxicological) doses. There are many well-characterized mechanisms for these dose-specific effects including signaling via single vs. multiple steroid receptors due to nonselectivity at higher doses (30), receptor down-regulation at high doses vs. up-regulation at low doses (151, 152), differences in the receptors present in various tissues (153, 154), cytotoxicity at high doses (155), and tissue-specific components of the endocrine-relevant transcriptional apparatus (104, 105). Some of these factors will be addressed in Section III.B in the section dedicated to NMDRCs.
F. Intrauterine position and human twins: examples of natural low-dose effects
Hormones have drastically different effects at different periods of development. In a now classical Endocrinology paper, Phoenix and colleagues (156) showed that hormone exposures during early development, and in particular fetal development, had organizational effects on the individual, whereby the developing organs were permanently reorganized by exposure to steroids. Permanent, nonreversible masculinization of the developing body plan by androgen exposure in utero is an example. These organizational effects are in contrast to the effects of the same hormones, at similar or even higher doses, on adults. The effects of steroids on individuals after puberty have been termed activational, because the effects on target organs are typically transient; withdrawal of the hormone returns the phenotype of the individual to the preexposed state (157), although this is not always the case (158).
One of the most striking examples of the ability of low doses of hormones to influence a large repertoire of phenotypes is provided by the study of intrauterine positioning effects in rodents and other animals. The rodent uterus in particular, where each fetus is fixed in position along a bicornate uterus with respect to its neighbors, is an excellent model to study how hormones released from neighboring fetuses (159) can influence the development of endocrine-sensitive endpoints (31). Importantly, differences in hormonal exposures by intrauterine position are relatively small (see Fig. 2) (160). Thus, even a small magnitude in differences of hormonal exposures is sufficient to generate effects on behavior, physiology, and development.

Intrauterine position produces offspring with variable circulating hormone levels. Fetuses are fixed in position in the bicornate rodent uterus, thus delivery via cesarean section has allowed for study of the influence of intrauterine position on behaviors, physiology, and organ morphology. Illustrated here are the differences in estradiol (E2) and testosterone (T) concentrations measured in male and female fetuses positioned between two male neighbors (2M), two female neighbors (2F), or neighbors of each sex (1MF). Direction of blood flow in the uterine artery (dark vessel) and vein (light vessel) is indicated by an arrow (159).
The earliest studies of intrauterine position compared behavioral characteristics of females relative to their position in the uterus (161–164); male behavior was also affected by intrauterine position (161, 165–167). Subsequent studies of intrauterine position showed that position in the uterus influenced physiological endpoints (157, 160–162, 168–174) as well as morphological endpoints in female rodents (160, 161, 163, 164, 175–177). Male physiology and morphological endpoints were similarly affected by intrauterine position (165, 167, 177–179).
The endocrine milieu of the uterine environment has been implicated in these effects because differences in hormonal exposure have been observed based on intrauterine position (Fig. 2). The production of testosterone in male mice starting at approximately d 12 of gestation allows for passive transfer of this hormone to neighboring fetuses (159, 160, 180). Thus, fetuses positioned between two male neighbors have slightly higher testosterone exposures compared with fetuses positioned between one male and one female or two female neighbors (168, 181–183). These data indicate that very small differences in hormone exposures during fetal development are capable of influencing a variety of endpoints, many of which become apparent only during or after puberty. Furthermore, small differences in hormone exposures may be compounded by other genetic variations such as those normally seen in human populations.
Intrauterine effects have been observed in animals with both large litters and singleton or twin births including ferrets, pigs, hamsters, voles, sheep, cows, and goats (136, 184, 185). But perhaps the most compelling evidence for intrauterine effects comes from human twin studies. Many studies have found that the sex of the fetuses impacts the phenotype of one or more of the twins, with significant evidence suggesting that male twins strongly influence a female co-twin; endpoints including sensation seeking (186), ear superiority (187, 188), brain and cerebellum volume (189), masculine/feminine behaviors and aggression levels (190–192), handedness (193, 194), reproductive fitness (192, 195), finger length ratios (196), risk for developing eating disorders (197), and birth weight (198) were all affected in females with a male twin. From these studies, many authors have concluded that testosterone from male fetuses influences developmental parameters in female twins; typically, male same-sex twins do not display altered phenotypes for these endpoints. Yet importantly, limited studies indicate that female twins can influence their uterine pairs, with some behaviors affected in male co-twins (191); breast cancer incidence in women and testicular cancer in men have also been shown to be influenced by having a female co-twin (83, 199, 200).
Although the mechanisms for these intrauterine effects are not completely understood, very small differences in hormone exposures have been implicated, making the effects of twin gestations a natural example of low-dose phenomena. In the human fetus, the adrenals produce androgens that are converted to estrogen by the enzyme aromatase, specifically in the placenta. In a human study designed to compare hormone levels in the amniotic fluid, maternal serum, and umbilical cord blood of singleton male and female fetuses, significant differences were observed in the concentrations of testosterone, androstenedione (A4), and estradiol (201). Specifically, amniotic fluid concentrations of testosterone and A4 were approximately twice as high in male fetuses, whereas estradiol concentrations were slightly, but significantly, higher in female fetuses. Yet, interestingly, there were no differences for any of the hormones in maternal serum, similar to findings in mice that litters with a high proportion of males or females did not impact testosterone, estradiol, or progesterone serum levels in mothers (180). In umbilical cord serum, concentrations of A4 and estradiol were higher in males compared with females (201), although it must be noted that these samples were collected at parturition, long after the fetal period of sexual differentiation of the reproductive organs.
Several studies have specifically compared steroid hormone levels in maternal and umbilical cord blood samples collected from same-sex and opposite-sex twins. Male twins, whether their co-twin was a male or a female, had higher blood concentrations of progesterone and testosterone compared with female twins (202). Furthermore, for both sexes, dizygotic twins had higher levels of these hormones, as well as estradiol, compared with monozygotic twins. Fetal sex had no effect on maternal concentrations of testosterone, progesterone, or estrogen, suggesting that any differences observed in fetal samples are due to contributions from the fetuses’ own endocrine systems and the placental tissue (203). Yet an additional study conducted in women carrying multiple fetuses (more than three) indicates that both estradiol and progesterone concentrations in maternal plasma increase with the number of fetuses, and when fetal reduction occurs, these hormone levels remain elevated (204).
It has been proposed that low-dose effects seen in different intrauterine positions in litter-bearing animals could be an evolutionary adaptation, whereby the genotypes of the fetuses are relatively similar but a range of phenotypes can be produced via differential hormone exposures (136, 168). For example, female mice positioned between two females are more docile and thus have better reproductive success when resources are plentiful, but females positioned between two males are more aggressive and therefore are more successful breeders under stressful conditions (161, 171, 175). In this way, a mother produces offspring with variable responses to environmental conditions, increasing the chances that her own genetic material will continue to be passed on. Yet although there is evidence to suggest that a variable intrauterine environment is essential for normal development (171), intrauterine positional effects appear to have little effect on offspring phenotypes in inbred rodent strains (168, 205). This result may be related to the link between genetic diversity and hormone sensitivity (206, 207), suggesting that outbred strains are the most appropriate for studying endocrine endpoints and are also most similar to the effects of low doses of hormones on human fetuses.
Finally, it has been proposed that similar mechanisms are used by the developing fetus in response to natural hormones via intrauterine position and EDCs with hormonal activity (136). To this end, several studies have examined the effects of both exposure to an EDC and intrauterine position or have considered the effect of intrauterine position on the response of animals to these chemicals (174, 176, 181, 208, 209). For example, one study found that intrauterine position affected the morphology of the fetal mammary gland, yet position-specific differences were obliterated by BPA exposure (176). Additional studies suggest that prostate morphology is disrupted by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure in males positioned between two females, but this chemical does not affect prostate morphology in males positioned between two males (181). Finally, male rodents positioned between two males have higher glucose intolerance than males positioned between two females, yet when these males are given a diet high in phytoestrogens, glucose tolerance is dramatically improved in the males positioned between two males, whereas their siblings positioned between two females do not benefit (209). What is clear from these studies is that low doses of natural hormones are capable of altering organ morphology, physiology, and reproductive development, similar to the effects of EDCs.
It has been suggested that the endocrine system allows for homeostatic control and that the aim of the endocrine system is to “maintain normal functions and development in the face of a constantly changing environment” (210). Yet studies from intrauterine position, together with studies of EDCs (see Sections II.C–F), clearly indicate that the fetal endocrine system cannot maintain a so-called homeostasis and is instead permanently affected by exposures to low doses of hormones.Go to:
II. Demonstrating Low-Dose Effects Using a WoE Approach
A. Use of a WoE approach in low-dose EDC studies
In 2001, the NTP acknowledged that there was evidence to support low-dose effects of DES, genistein, methoxychlor, and nonylphenol (2). Specifically, the NTP expert panel found that there was sufficient evidence for low-dose effects of DES on prostate size; genistein on brain sexual dimorphisms, male mammary gland development, and immune responses; methoxychlor on the immune system; and nonylphenol on brain sexual dimorphisms, thymus weight, estrous cyclicity, and immune responses. Using the NTP’s definitions of low dose (i.e. effects occurring in the range of typical human exposures or occurring at doses lower than those typically used in standard testing protocols), we propose that most if not all EDCs are likely to have low-dose effects. Yet an important caveat of that statement is that low-dose effects are expected for particular endpoints depending on the endocrine activity of the EDC, and not for any/all endocrine-related endpoints. For example, if a chemical blocks the synthesis of a hormone, blood levels of the hormone are expected to decline, and the downstream effects should then be predicted from what is known about the health effects of low hormone levels. In contrast, if a chemical binds a hormone receptor, the effects are expected to be very complex and to be both tissue specific and dose specific. Finally, most EDCs interact with multiple hormone pathways, or even multiple hormone receptors, making the expected effects even more complex and context specific (211–213).
Table 3 summarizes a limited selection of chemicals that have evidence for low-dose effects, with a focus on in vivo animal studies. As seen by the results presented in this table, low-dose effects have been observed in chemicals from a number of classes with a wide range of uses including natural and synthetic hormones, insecticides, fungicides, herbicides, plastics, UV protection, and other industrial processes. Furthermore, low-dose effects have been observed in chemicals that target a number of endocrine endpoints including many that act as estrogens and antiandrogens as well as others that affect the metabolism, secretion, or synthesis of a number of hormones. It is also clear from this table that the cutoff for low-dose effects is not only chemical specific but also can be effect dependent. And finally, although this table is by no means comprehensive for all EDCs or even the low-dose effects of any particular chemical, the affected endpoints cover a large range of endocrine targets.
Table 3.
EDCs with reported low-dose effects in animals (or humans, where stated)
| Chemical | Use | EDC action | Low-dose cutoff | Affected endpoint | Refs. |
|---|---|---|---|---|---|
| Aroclor 1221 (PCB mixture) | Coolants, lubricants, paints, plastics | Mimics estrogens, antiestrogenic activity, etc. | 0.1–1 mg/kg (produces human blood levels) | Brain sexual dimorphisms | 683, 684 |
| Atrazine | Herbicide | Increases aromatase expression | 200 μg/liter (334, 335) | Male sexual differentiation/development | See this review |
| BPA | Plastics, thermal papers, epoxy resins | Binds ER, mER, ERRγ, PPARγ, may weakly bind TH receptor and AR | 400 μg/kg · d (produces human blood concentrations) | Prostate, mammary gland, brain development and behavior, reproduction, immune system, metabolism | See this review |
| Chlordane | Insecticide | Binds ER | 100 ng/g (produces human blood levels) | Sexually dimorphic behavior | 685 |
| Chlorothalonil | Fungicide, wood protectant | Aromatase inhibitor | 164 μg/liter (environmental concentrations, EPA) | Corticosterone levels (amphibians) | 686 |
| Chlorpyrifos | Insecticide | Antiandrogenic | 1 mg/kg · d (EPA) | Acetylcholine receptor binding (brain) | 687 |
| DDT | Insecticide | Binds ER | 0.05 mg/kg (EPA) | Neurobehavior | 688 |
| DES | Synthetic hormone | Binds ER | 0.3–1.3 mg/kg · d (dose typically administered to pregnant women) | Prostate weight | 689 |
| Dioxin (TCDD) | Industrial byproduct | Binds AhR | 1 μg/kg · d (397) | Spermatogenesis, immune function and oxidative stress, tooth and bone development, female reproduction, mammary gland, behavior | See this review |
| Genistein | Phytoestrogen | Binds ER | 50 mg/kg (EPA) | Brain sexual dimorphisms | 690 |
| Heptachlor | Insecticide | Induces testosterone hydroxylases | 0.15 mg/kg · d (EPA) | Immune responses | 691 |
| Hexachlorobenzene | Fungicide | Modulates binding of ligand to TRE, weakly binds AhR | 0.08 mg/kg · d (EPA) | Anxiety and aggressive behaviors | 692 |
| Maneb | Fungicide | Inhibits TSH release, may bind PPARγ | 5 mg/kg · d (EU Commission) | Testosterone release | 693 |
| Methoxychlor | Insecticide | Binds ER | 5 mg/kg · d (WHO) | Immune system | 694, 695 |
| 4-Methylbenzylidine camphor | UV screen | Weakly estrogenic | 10 mg/kg · d (Europa) | Sexual behavior | 696 |
| Methyl paraben | Preservative | Estrogenic | 1000 mg/kg · d (EFSA) | Uterine tissue organization | 697 |
| Nicotine | Natural alkaloid in tobacco | Binds acetylcholine receptors, stimulates epinephrine | Human use of nicotine substitutes | Incidence of cryptorchidism (humans) | 698 |
| Nonylphenol | Detergents | Weakly estrogenic | 15 mg/kg · d (EPA) | Testosterone metabolism | 699 |
| Octylphenol | Rubber bonding, surfactant | Weakly binds ER, RXR, PRGR | 10 mg/kg · d (700) | Testes endpoints | 701 |
| Parathion | Insecticide | 0.2 mg/kg · d (WHO) | Cognitive and emotional behaviors | 702 | |
| PBDE-99 | Flame retardant | Alters TH synthesis | 0.3 mg/kg · d (EPA) | TH levels in blood | 703 |
| PCB180 | Industrial lubricant, coolant | Impairs glutamate pathways, mimics estrogen | Examined normal human populations | Diabetes (humans) | 704 |
| PCB mixtures | Coolants, lubricants, paints, plastics | Binds AhR, mimic estrogens, antiestrogenic activity, etc. | Each at environmentally relevant levels | TH levels | 705 |
| Perchlorate | Fuel, fireworks | Blocks iodide uptake, alters TH | 0.4 mg/kg · d (436) | TSH levels (humans) | See this review |
| Sodium fluoride | Water additive (to prevent dental caries), cleaning agent | Inhibits insulin secretion, PTH, TH | 4 mg/liter water (EPA standard) | Bone mass and strength | 706 |
| Tributyltin oxide | Pesticide, wood preservation | Binds PPARγ | 0.19 mg/kg · d (EPA) | Obesity | 707 |
| Triclosan | Antibacterial agent | Antithyroid effects, androgenic and estrogenic activity | 12 mg/kg · d (Europe SCCP) | Altered uterine responses to ethinyl estradiol | 708 |
| Vinclozolin | Fungicide | Antiandrogenic | 1.2 mg/kg · d (EPA) | Male fertility | 709 |
EDC action indicates that for some chemicals, an effect is observed (i.e. estrogenic, androgenic), but for many EDCs, complete details of receptor binding are unavailable or incomplete. Low-dose cutoff means the lowest dose tested in traditional toxicology studies, or doses in the range of human exposure, depending on the data available. Affected endpoint means at least one example of an endpoint that shows significant effects below the low-dose cutoff dose. This list is not comprehensive, and the lack of an endpoint on this table does not suggest that low doses do or do not affect any other endpoints. AR, Androgen receptor; EFSA, European Food Safety Authority; ERR, estrogen related receptor; PCB, polychlorinated biphenyl; PPARγ, peroxisome proliferator-activated receptor-γ; PRGR, progesterone receptor; RXR, retinoid X receptor; SCCP, Scientific Committee on Consumer Products; TH, thyroid hormone; TRE, thyroid response element; WHO, World Health Organization.
Several EDCs have been well studied, and the number of publications focusing on low-dose effects on a particular developmental endpoint is high; however, other chemicals are less well studied with fewer studies pointing to definitive low-dose effects on a given endpoint. In fact, there are a significant number of EDCs for which high-dose toxicology testing has been performed and the no observed adverse effect level (NOAEL) has been derived, but no animal studies in the low-dose range have been conducted, and several hundred additional EDCs where no significant high- or low-dose testing has been performed (see Table 4 for examples). Balancing the large amount of data collected from some well-studied chemicals like BPA and atrazine with the relative paucity of data about other chemicals is a difficult task.
Table 4.
Select examples of EDCs whose potential low-dose effects on animals remain to be studied
| Chemical | Use | EDC action | Low-dose cutoff |
|---|---|---|---|
| Antiseptics and preservatives | |||
| Butyl paraben | Preservative (cosmetics) | Estrogenic, antiandrogenic | 2 mg/kg · d (EPA) |
| Propyl paraben | Antimicrobial preservative found in pharmaceuticals, foods, cosmetics, and shampoos | Estrogenic activity | LOAEL 10 mg/kg · d, NOEL 6.5 mg/kg · d (Europa) |
| Cosmetics and personal care products | |||
| 2,4-Dihydroxybenzophenone | UV absorber in polymers, sunscreen agent | Estrogenic activity | Not identified |
| 3-Benzylidene camphor | UV blocker used in personal care products | Estrogenic activity | 0.07 mg/kg · d (710) |
| 4,4′-Dihydroxybenzophenone | UV light stabilizer used in plastics, cosmetics, adhesives, and optical fiber | Estrogenic activity | Not identified |
| Benzophenone-2 | Used in personal care products such as aftershave and fragrances | Estrogenic activity, changes in T4, T3, and TSH levels, alterations in cholesterol profile | NOEL 10–333 mg/kg · d (711) |
| Benzophenone-3 | UV filter | Estrogenic, PPARγ activator | 200 mg/kg · d (Europa) |
| Multiple use (other) | |||
| Melamine | Flame-retardant additive and rust remover; used to make laminate, textile, and paper resins; metabolite of cyromazine | Affects voltage-gated K+ and Na+ channels and Ca2+ concentrations in hippocampal neurons | 63.0 mg/kg · d (FDA) |
| Resorcinol | Used in the manufacturing of cosmetics, dyes, flame retardants, hair dye formulations, pharmaceuticals, skin creams, and tires | Alters T4 and TSH levels | 80.00 mg/kg · d (Europa) |
| Pesticides | |||
| Aldrina | Insecticide | Estrogenic activity | 0.025 mg/kg · d (Health Canada) |
| Alachlor | Herbicide | Decreases serum T4, binds PR, weakly binds ER | 1 mg/kg · d (EPA) |
| Amitrole | Herbicide | Decreases thyroid hormone | 0.12 mg/kg · d (FAO) |
| Bitertanol | Fungicide | Alters aromatase | 30 mg/kg · d (EPA) |
| Carbendazim | Fungicide | Affects FSH, LH, and testosterone levels; alters spermatogenesis and Sertoli cell morphology | 8 mg/kg · d (712) |
| Diazinon | Insecticide | Alters glucocorticoids | 0.065 mg/kg · d (CDC) |
| Endrina | Insecticide | Stimulates glucocorticoid receptor | 0.025 mg/kg · d (CDC) |
| Fenoxycarb | Insecticide | Alters acetylcholinesterase | 260 mg/kg · d (CDC) |
| Mirexa | Insecticide | Decreases testosterone levels | 0.075 mg/kg · d (CDC) |
| Zineb | Fungicide | Alters T4 and dopamine levels | LOAEL 25 mg/kg · d (EPA) |
| Ziram | Fungicide | Alters norepinephrine levels | 1.6 mg/kg · d (EPA) |
| Resins | |||
| Bisphenol F | Used in polycarbonates | Alters T4, T3, and adiponectin levels, has estrogenic activity | LOAEL 20 mg/kg · d (713) |
| Styrene | Precursor to polystyrene | Alters dopamine | 200 mg/kg · d (EPA) |
PPARγ, peroxisome proliferator-activated receptor-γ; PR, progesterone receptor.aThese chemicals were identified in the 1990s as part of the dirty dozen, 12 chemicals that were acknowledged to be the worst chemical offenders because of their persistence in the environment, their ability to accumulate through the food chain, and concerns about adverse effects of exposures to wildlife and humans. These chemicals were banned by the Stockholm convention and slated for virtual elimination. Yet there is still very little known about the low-dose effects of these chemicals, likely in the range of past and current human and/or wildlife exposures.
WoE approaches have been used in a large number of fields to determine whether the strength of many publications viewed as a whole can provide stronger conclusions than any single study examined alone. Although the term ‘weight of evidence’ is used in public policy and the scientific literature, there is surprisingly little consensus about what this term means or how to characterize the concept (214). Historically, risk assessors have used qualitative approaches (i.e. professional judgment to rank the value of different cases) and quantitative approaches (i.e. scoring methods to produce statistical and mathematical determinations of chemical safety), but it has been argued that these methods lack transparency and may produce findings that are unrepeatable from one risk assessor to another (215, 216). Whatever the method used, when EDCs are being assessed, it is important to use the principles of endocrinology to establish the criteria for a WoE approach. We do this in Section II.B, identifying three key criteria for determining whether a study reporting no effect should be incorporated into a WoE approach. It also should be noted that in epidemiology, the term ‘weight of evidence’ is typically not used, but the concept is actuated by meta-analysis, formally and quantitatively combining data across studies, including a plot of individual and pooled study findings and also a measure of heterogeneity of findings between studies.
For some well-studied chemicals, there are large numbers of studies showing both significant effects, and additional studies showing no effects, from low-dose exposures. In these cases, extensive work is needed to deal with discordant data collected from various sources; studies showing no effect of low-dose exposures must be balanced in some way with those studies that do show effects. As stated by Basketter and colleagues (217), “it is unwise to make a definitive assessment from any single piece of information as no individual assay or other assessment … is 100% accurate on every occasion … This means that from time to time, one piece of conflicting data has to be set aside.” WoE approaches in EDC research have typically dealt with datasets that have some conflicting studies, and these conflicts are even more difficult to sort out when studies have attempted to directly replicate published findings of adverse effects (see for example Refs. 218–221).
Most previously published WoE analyses have examined chemicals broadly (asking questions such as, “Does BPA produce consistent adverse effects on any endpoint?”) (see Ref. 222). This can lead to problems including those encountered by the NTP expert panel, which found that there was some evidence for low-dose effects of BPA on certain endpoints but mixed findings for other endpoints. For example, the panel noted that some studies found low-dose effects of BPA on the prostate, but other studies could not replicate these findings. In Section II.B, we address criteria that are needed to accept those studies that are unable to detect low-dose effects of chemicals; these criteria were not used by the NTP in 2001, but they are essential to address controversies of this sort and perform WoE analyses using the best available data. In the sections that follow, we employed a WoE approach to examine the evidence for low-dose effects of single chemicals on selected endpoints or tissues, also paying attention to when in development the EDCs in question were administered.
B. Refuting low-dose studies: criteria required for acceptance of studies that find no effect
Over the past decade, a variety of factors have been identified as features that influence the acceptance of low-dose studies (69, 71, 76, 77, 90, 205, 223, 224). In fact, the NTP low-dose panel itself suggested that factors such as strain differences, diet, caging and housing conditions, and seasonal variation can affect the ability to detect low-dose effects in controlled studies (2). In particular, three factors have been identified; when studies are unable to detect low-dose effects, these factors must be considered before coming to the conclusion that no such effects exist.
1. Negative controls confirm that the experimental system is free from contamination
Although all scientific experiments should include negative (untreated) controls, this treatment category is particularly important for EDC research. When a study fails to detect low-dose effects, the observed response in control animals should be compared with historical untreated controls; if the controls deviate significantly from typical controls in other studies, it may indicate that these animals were, in fact, treated or contaminated in some way or that the endpoint was not appropriately assessed (77, 205, 225). For example, if an experiment was designed to measure the effect of a chemical on uterine weight, and the control uteri have weights that are significantly higher than is normally observed in the same species and strain, these animals may have been inadvertently exposed to an estrogen source, or the uteri may not have been dissected properly by the experimenters. In either case, the study should be examined carefully and likely cannot be used to assess low-dose effects; of course, untreated controls should be monitored constantly because genetic drift and changes in diet and housing conditions can also influence these data, thus explaining changes from historical controls. Importantly, several types of contamination have been identified in studies of EDCs including the leaching of chemicals from caging or other environmental sources (226, 227), the use of pesticide-contaminated control sites for wildlife studies and contaminated controls in laboratory studies (76), and even the use of food that interferes with the effects of EDCs (224, 228). It is also important to note that experiments must consider the solvent used in the administration of their test chemical, and thus good negative controls should test for effects of the solvent itself. Using solvent negative controls helps prevent false positives as well as the possibility that the vehicle could mask the effects of the chemical being studied.
2. Positive controls indicate that the experimental system is capable of responding to low doses of a chemical acting on the same pathway
Many studies do not include a positive control, either because of the size and cost of the experiment when including an additional treatment or because an appropriate positive control has not been identified for the endpoint being examined. If the experiment detects an effect of the chemical in question, the exclusion of a positive control does not necessarily affect the interpretation of the results; instead, it can be appropriately concluded that the test chemical is significantly different from unexposed (but similarly handled/treated) negative controls. However, if the study fails to detect low-dose effects of a test chemical, no convincing conclusion can be made; in this case, a positive control is required to demonstrate that the experimental system was capable of detecting such effects (71, 75, 77, 205).
Several issues must be considered when addressing whether the positive control confirms the sensitivity of the assay. First, an appropriate chemical must be selected, and it must be administered via the appropriate route, i.e. if the test chemical is administered orally, a positive control that is orally active, such as ethinyl estradiol, should be used; if the test chemical is administered sc, a positive control that is active via this route, such as 17β-estradiol, is most appropriate. The use of 17β-estradiol in studies that use oral exposures is particularly inappropriate (see Ref. 229) for example) because this hormone, like most natural steroids, has very low oral activity (77). Second, the positive control chemical must be examined, and effective, at appropriately low doses. Thus, if the test chemical is 100 times less potent than the positive control, a dose of the positive control 100 times lower than the test compound must produce effects (69, 71, 205). For example, studies that report effects of ethinyl estradiol only at doses that are hundreds of times higher than the dose that is effective in contraceptives (230) are not capable of detecting low-dose effects of test chemicals. Without appropriate and concurrent positive and negative controls, studies that fail to detect low-dose effects of test chemicals should be rejected.
3. Species and animal strains that are responsive to EDCs must be used
The NTP expert panel specifically noted that “because of clear species and strain differences in sensitivity, animal-model selection should be based on responsiveness to endocrine-active agents of concern (i.e. responsive to positive controls), not on convenience and familiarity” (2). An analysis of the BPA literature clearly showed that many of the studies that failed to detect effects of low doses used the Charles River Sprague-Dawley rat (75); this strain was specifically bred to have large litters (231), and many generations of inbreeding have rendered the animal relatively insensitive to estrogens (205). The NTP expert panel noted the lack of effects of BPA on Sprague-Dawley rats and concluded that there were clear differences in strain sensitivity to this chemical (2). Importantly, this may not be true for Sprague-Dawley rats that originate from other vendors, indicating that animal origin can also influence EDC testing.
Many studies in mice (138, 206, 207, 232–234) and rats (232, 235–239) have described differences displayed between two (or more) animal strains to a natural hormone or EDC. Often these differences can be traced to whether a strain is inbred or outbred. Genetically diverse strains are generally found to be more sensitive to estrogens (206). Importantly, well-controlled studies demonstrate that strain differences in response to estrogen treatment may be organ dependent or may even differ between levels of tissue organization within the same organ. For example, the Sprague-Dawley rat is more sensitive to ethinyl estradiol than other strains when measured by uterine wet weight. However, when other endpoints were measured, i.e. height of cells in the uterine epithelium, the Sprague-Dawley rat was indistinguishable from the DA/Han rat; instead, the Wistar rat had the most heightened response (237). Additionally, there are data to indicate that strain differences for one estrogen may not be applicable for all estrogenic chemicals. In comparing the responses of DA/Han, Sprague-Dawley, and Wistar rats to other xenoestrogens, additional differences were observed including a greater increase in uterine wet weight of DA/Han and Sprague-Dawley rats but not Wistar rats after exposure to 200 mg/kg BPA; increased uterine epithelium thickness was observed in Wistar and Sprague-Dawley rats but not DA/Han rats after exposure to 200 mg/kg octylphenol (237). Attempts have been made, at times successfully, to map the differences in strain response to genetic loci (240). However, it appears that strains with differences in response that manifest in some organs do not have divergent responses in other organs, a phenomenon that is not explained by genetic differences alone. For these reasons, the NTP’s recommendation that scientists use animals that are proven responsive to EDCs (2) must be observed.
4. Additional factors?
Additional factors have also been identified as influential in the ability (or inability) to detect low-dose effects in EDC studies. Although these factors must be considered when interpreting studies and using a WoE approach, some issues that were previously identified as essential factors in the design of studies (i.e. route of administration) have more recently been disputed (241).
The first factor is the use of good laboratory practices (GLP) in the collection of data. When assessing the EDC literature for risk assessment purposes, the FDA and European Food Safety Authority (EFSA) have given special prominence to studies that complied with GLP guidelines, essentially giving scientific priority to industry-funded studies because that group typically conducts GLP guideline studies (33, 242). Because GLP guidelines are designed only to control data collection, standards for animal care, equipment, and facility maintenance, and they do not ensure that studies were designed properly with the appropriate controls, it has been argued that the use of GLP methods is not appropriate or required for EDC studies (69).
GLP studies are typically large, with dozens of animals studied for each endpoint and at each time point. Thus, it has been concluded that these studies are better simply because they are larger. Yet small studies designed with the use of power analysis, statistical tools that allow researchers to determine a priori the number of animals needed to determine significant differences based on effect size, are equally capable of detecting effects while reducing the number of animals used (69). GLP studies also typically (but not necessarily) rely upon standardized assays, which are not generally considered contemporary tools and are often shown to be incapable of detecting adverse effects on endpoints that employ modern tools from molecular genetics and related disciplines. Furthermore, some fields of EDC research have no GLP studies (243). Finally, there is no published evaluation of whether studies performed under GLP are more capable of providing accurate results. The priority given to GLP studies therefore does not appear to have been justified based on any comparative analysis. Thus, as long as studies include appropriate measures of quality assurance, they need not be performed under GLP standards to provide reliable and valuable information, and many GLP studies are inadequate to assess important and relevant endpoints. Instead, the most valuable studies consider the factors presented above, along with appropriate dose selections and choice of endpoint.
The second factor worth considering is the source of funding for studies. In several fields, significant controversy has been produced based on the results obtained from independent scientists compared with results obtained from scientists affiliated with the chemical industry (75, 76). Funding source per se should not dictate the outcome of a research study, but that does not mean that researchers are not subject to underlying biases. In our own WoE analyses, presented in Sections II.C–G, we do not discount studies merely because they were conducted with industry funds, nor do we lend higher weight to studies conducted in independent or government laboratories; if a study, regardless of funding, finds no effect of a chemical, it is given weight only if the three criteria described in Sections II.B.1–3 (successful and appropriate negative and positive controls and appropriate choice of animal model) were met.
To perform a WoE evaluation, we identified some basic information about the chemical in question, the dose that would be considered a low-dose cutoff, and the studies in support of and against low-dose effects. We then considered whether the majority of studies found effects of low doses of a chemical on a single endpoint in question. If studies did not find low-dose effects, we considered whether they adhered to the criteria discussed above for proper design of an EDC low-dose study. In particular, we considered whether appropriate animal strains as well as positive and negative controls were used. With regard to animal strain, as discussed briefly in Section II.B.3, there is variability between animal strains that can significantly influence the ability to detect effects of EDCs; using insensitive strains to produce negative data cannot refute positive data in a sensitive strain. In several cases, it was easy to conclude that there was a strong case for low-dose effects because there were no studies finding no effects at low doses or because all of the negative studies were inappropriately designed. For other chemicals, a significant number of studies found effects on the endpoint being considered, but other (adequately designed) studies refuted those findings. Under those circumstances, we determined whether the findings of harmful effects came from multiple laboratories; when they did, we cautiously concluded that there was evidence for low-dose effects. Below (Sections II.C–G), we present five examples where a significant number of studies were available examining low-dose effects of an EDC on a single particular endpoint.
C. BPA and the prostate: contested effects at low doses?
As discussed briefly above, BPA is one of the best-studied EDCs, with more than 200 published animal studies, many of which focused on low doses (29, 31). The effects of this chemical on wildlife species have also been described in detail (28). BPA is found in a myriad of consumer products, and it leaches from these items under normal conditions of use (4). It has also been regularly detected in air, water, and dust samples. The majority of individuals in industrialized countries have BPA metabolites in their urine, and trends indicate increasing exposures in developing nations like China (87, 244). Although it was long suspected that most human exposures originate from BPA contamination of food and beverages, a study comparing the excretion of BPA metabolites with the length of time spent fasting suggests that there are also likely to be significant exposures from sources other than food and beverages (245). BPA has recently been shown to be used in large quantities in thermal and recycled papers and can enter the skin easily via dermal absorption (246–248). Thus, despite the large amount of information available on BPA sources, our understanding of how these sources contribute to total human exposures remains poor; these studies also point to significant gaps in current knowledge about BPA metabolism in humans (243).
BPA binds to the nuclear and membrane ER, and thus most of the effects of this chemical have been attributed to its estrogenic activity (27). However, there is evidence that it can activate a number of additional pathways, including thyroid hormone receptor, androgen receptor, as well as peroxisome proliferator-activated receptor-γ signaling pathways (249–252). The cutoff for a low dose has been set at several different concentrations depending on which studies and definitions are used (see Table 1). The EPA calculated a reference dose for BPA of 50 μg/kg · d based on a LOAEL of 50 mg/kg · d (38). More recent pharmacokinetic scaling experiments have estimated that exposures to approximately 400 μg/kg · d produce blood concentrations of unconjugated BPA in the range of human blood concentrations (4). Thus, for the two WoE analyses of the BPA literature we conducted, doses of 400 μg/kg · d or lower were considered low dose; pharmacokinetic studies from nonhuman primates support the appropriateness of this dose for approximating human exposure levels (253). Furthermore, because this dose is below the toxicological LOAEL, it is a conservative cutoff for low-dose studies (see Refs. 3 and 38 and Table 1).
One of the most well studied and hotly debated examples of a low-dose effect comes from the BPA literature; regulatory agencies and scientists have addressed several times whether low doses of BPA during fetal and perinatal development affect the rodent prostate (118, 205, 254, 255). In 1997, the first study on BPA and the prostate determined that fetal exposure to low doses (2 and 20 μg/kg · d administered orally to pregnant mice) increased the weight of the adult prostate compared with unexposed male offspring (256). Since that time, several additional studies have verified that prostate weight is affected by fetal exposure to similar low doses (257–259). Studies have also shown that low doses of BPA affect androgen receptor binding activity in the prostate (257), tissue organization, and cytokeratin expression in the gland (260–262) as well as the volume of the prostate and the number and size of dorsolateral prostate ducts (208). Several recent studies have also examined whether low doses of BPA (10 μg/kg · d) influence the incidence of adult-onset prostatic intraepithelial neoplasia (PIN) lesions. Perinatal BPA exposure, whether administered orally or sc to pups, increases the incidence of PIN lesions in response to a mixture of testosterone and estradiol in adulthood (139, 141, 263); this hormonal cocktail was designed to mimic the endocrine changes associated with aging in men that also typically accompany the onset of prostate cancer. In addition to the effects of BPA on PIN lesions, these low doses also produced permanent alterations in the epigenome of exposed males, with prostates displaying completely unmethylated sequences in genes that are hypermethylated in unexposed controls (140, 263). In examining these studies, although the same effects of BPA on the prostate were not observed in all studies, there is an obvious trend demonstrating that low doses of BPA during early development significantly affect several aspects of prostate development.
Since the initial report showing effects of low doses on the prostate, approximately nine studies, including several designed specifically to replicate the original positive study, have shown no effects of low doses on the prostate (264–272); every one of these studies examined the prostate weight, and Ichihara et al. (264) also examined the effects of BPA on PIN lesions (without hormonal treatment) and the response of the prostate to a chemical carcinogen. Three of these studies failed to include a positive control of any kind (264, 268, 270); three studies used DES as a positive control but found no effect from exposure to this potent xenoestrogen (265–267) (i.e. the positive control failed); another study used 17β-estradiol as a positive control, inappropriately administered orally, and found no effects of this hormone on the prostate (271); and two studies used an estrogenic positive control (ethinyl estradiol) and found effects from its exposure, but only at inappropriately high doses (269, 272). These two studies clearly showed that the positive control dose was too high, because rather than increase the weight of the prostate (as seen after low doses of estrogens in other studies), the positive control decreased the weight of the adult prostate (269, 272).
Although this topic was once considered controversial, using a WoE approach, it is clear that there is strong evidence in support of low-dose effects of BPA on the development of the prostate. The evidence clearly shows that several endpoints, including prostate weight, were affected in similar ways in multiple studies from several different labs at doses below 400 μg/kg · d; most effects were seen at doses below 50 μg/kg · d. Furthermore, PIN lesions were reported after neonatal exposure to 10 μg/kg · d with hormonal treatment in adulthood. No appropriately conducted studies contest this evidence. Therefore, the WoE analysis demonstrates that low doses of BPA significantly alter development of the rodent prostate. The NTP’s review of the BPA literature in 2008 indicated that this agency agrees that there is now significant evidence that low-dose BPA adversely affects development of the prostate (273).
D. BPA and the mammary gland: undisputed evidence for low-dose effects
The mammary gland is a conspicuous choice to examine the effects of estrogenic compounds because this organ depends on estrogen for proper development at several critical periods in life (274). The fetal gland expresses ER in the mesenchymal compartment, and just before birth, the epithelium becomes ER positive as well (275). At puberty, estrogen is responsible for ductal elongation and overall development of the gland, allowing the epithelium to fill the stromal compartment in preparation for pregnancy and lactation. Although BPA is an example of a chemical that has been classified as a weak estrogen because it binds with a much lower affinity to ERα compared with 17β-estradiol, even weak estrogens are known to affect the development of the mammary gland during early development (276).
In the first study to examine the effects of BPA on the mammary gland, prepubertal rats were exposed to relatively high doses (100 μg/kg · d or 54 mg/kg · d) for 11 d. After even this short exposure, mammary gland architecture was affected in both dose groups, with increased numbers of epithelial structures and, in particular, structures that suggest advanced development (277). BPA exposure also altered proliferation rates of mammary epithelium and cell cycle kinetics, with an increased number of cells in S-phase and a decreased number of cells in G1. Although relatively high doses of BPA were examined, this initial study indicated that the prepubertal and pubertal gland could be sensitive to BPA.
Many additional studies have examined another critical period, the fetal and neonatal periods, which are sensitive to environmental estrogens (78, 276, 278). Mice exposed prenatally to low doses of BPA via maternal treatment (0.25 μg/kg · d) displayed altered development of both the stromal and epithelial compartments at embryonic d 18, suggesting that exposures affect tissue organization during the period of exposure (176). In addition, similar low doses produced alterations in tissue organization observed in puberty and throughout adulthood, long after exposures ended, and even induced pregnancy-like phenotypes in virgin females (137, 279–282). Female mice exposed to BPA in utero displayed heightened responses to estradiol at puberty, with altered morphology of their glands compared with animals exposed to vehicle in utero (138). Another study demonstrated that perinatal BPA exposure altered the mammary gland’s response to progesterone (283). Remarkably, all of these effects were observed after maternal exposures to low doses (0.025–250 μg/kg), suggesting that the gland is extremely sensitive to xenoestrogen exposures. These studies are in contrast to one that examined the effects of higher doses (0.5 and 10 mg/kg · d) when BPA was administered for 4 d to the dam, which reported advanced development of BPA-exposed glands before puberty but no effects in adulthood (284).
Adult exposure to BPA is only now being examined in the mouse mammary gland model. A recent study examined the effects of BPA on mice with mutations in the BRCA1 gene. This study reported that 4 wks of exposure to a low dose of BPA altered the tissue organization of the mammary gland in ways that are similar to the effects observed after perinatal exposure (285). This study focused on altered development of the gland during exposure; additional studies are needed to determine whether these effects are permanent or whether normal mammary morphology could be achieved by cessation of BPA exposure.
Another obvious endpoint is the effect of BPA exposure on mammary cancer incidence. Several studies indicate that exposure to BPA in utero produces preneoplastic (281, 286, 287) and neoplastic lesions (286) in the gland in the absence of any other treatment. Additionally, other studies show that females exposed to BPA during the perinatal period are more sensitive to mammary carcinogens, decreasing tumor latency and increasing tumor incidence (287–290). These studies are also supported by subsequent studies examining gene and protein expression, which show that low-dose BPA specifically up-regulates expression of genes related to immune function, cell proliferation, cytoskeletal function, and estrogen signaling and down-regulates apoptotic genes (282, 288, 289, 291).
Postnatal BPA exposures also influence mammary cancer incidence; animals exposed lactationally to BPA from postnatal d 2 until weaning displayed decreased tumor latency and increased tumor multiplicity after treatment with DMBA [7,12-dimethylbenz(a)anthracene], a carcinogen (292). This study suggested that BPA exposure led to increased cell proliferation and decreased apoptosis in the gland and shifted the period where the gland is most susceptible to mammary carcinogens, a result that has important implications for human breast cancer. Finally, an additional study examined the effects of adult BPA exposure on mammary cancer; this study demonstrated that low doses of BPA accelerate the appearance of mammary tumors in a tumor-prone mouse strain (293). Interestingly, high doses did not have this effect; thus, this study is also an excellent example of a NMDRC.
Two studies of BPA and the mammary gland seem to contradict this body of literature, but both examined extremely high doses. In the first study, Nikaido et al. (294) exposed female mice to 10 mg/kg BPA from postnatal d 15–18. Mammary glands from these animals were examined at 4, 8, and 24 wk of age, and no differences were observed in the exposed animals relative to controls. Although the lack of effects reported in this study could be due to the high dose employed, they could also be related to the relatively short exposure period during the preweaning phase. In the second study, Yin and colleagues (295) examined the effects of BPA during the first few days after birth (0.1 or 10 mg BPA, equivalent to approximately 10 and 1000 mg/kg) on the incidence of mammary tumors after exposure to a mammary carcinogen at puberty. Similar to the study described above, this one also examined the effects of BPA after a relatively short period of exposure (only three injections administered between postnatal d 2 and 6). Although the study showed that BPA affected tissue organization, there was no change in the incidence of tumors in BPA-exposed females. Because both of these studies examined both high doses and relatively short periods of exposure, it is difficult to compare them directly to the studies finding effects of BPA on the mammary gland after longer exposures to lower doses; at the very least, they cannot refute studies suggesting that BPA alters development of this gland.
In summary, the WoE clearly shows that low-dose BPA exposure affects development of the mammary gland, mammary histogenesis, gene and protein expression in the gland, and the development of mammary cancers. In fact, this example of low-dose effects produced remarkably similar effects across more than a dozen studies conducted in several different labs. These results are also consistent with the effects of low-dose BPA exposure on mammary epithelial cells in culture (reviewed in Ref. 30). Although epidemiology studies examining the influence of BPA on breast cancer rates have proven to be inconclusive at best (296), to replicate the animal studies discussed above, epidemiologists must collect information about prenatal and neonatal exposures and relate them to adult breast cancer incidence. These types of studies would take decades to conduct (67) and should take into consideration the effects of other estrogens, because their effects can be additive or even synergistic (143, 144, 297).
Although our analyses of BPA have focused on its effects on the mammary gland and prostate (see Sections II.C–D), it is worth noting that several other endpoints have strong data to support the hypothesis that BPA has low-dose effects. In a recent review using similar WoE approaches, Hunt and colleagues (298) focused on those studies that examined the effects of BPA on the oocyte, specifically scrutinizing studies that reported effects, or no effects, on meiotic aneuploidy and other alterations in the intracellular organization and chromosome abnormalities. Similar to what has been observed with the prostate and mammary gland, the effects observed in the oocyte are variable from study to study, but overall consistent, and suggest that BPA exposure produces defects in these cells.
A large number of studies have also focused on the effects of BPA on the brain and behavior, with the most significant effects on sexually dimorphic regions of the brain and behaviors (299–307). Other affected behaviors include social behaviors, learning and anxiety, and maternal-neonate interactions (reviewed in Refs. 29 and 308). The NTP expert panel statement concluded that there were significant trends in these behavioral data and wrote that there was some concern that BPA could have similar effects in humans (273). Low-dose effects have also been reported for BPA in the female reproductive tract (309, 310), immune system (311, 312), maintenance of body weight and metabolism (313, 314), fertility (315–317), and the male reproductive tract (259, 318) (see Refs. 29 and 319 for comprehensive reviews).
E. Another controversial low-dose example: atrazine and amphibian sexual development
Atrazine is an herbicide that is applied in large volumes to crops, and there is concern that agricultural runoff of this chemical can affect nontarget animal species, especially amphibians that live and reproduce in small ponds and streams where significant amounts of atrazine have been regularly measured (320–322). It is the most commonly detected pesticide in ground and drinking water. Atrazine induces aromatase expression in cells and animals after exposure (323); this ultimately causes an increase in the conversion of testosterone to estrogen (324, 325). This effect has been reported in all vertebrate classes examined: fish, amphibians, reptiles, birds, and mammals, including human cell lines (see Ref. 326 for review). Another well-documented effect of atrazine is that it decreases androgen synthesis and activity, again, in every vertebrate class examined (326). In addition, endocrine-disrupting effects of atrazine occur through a number of other mechanisms, including antiestrogenic activity (327), altered prolactin release (328), and increased glucocorticoid release from the adrenal glands (329, 330), among others (327).
Because of atrazine’s indirect effect on estrogen levels, one relevant endpoint that has been given attention is the effect of this chemical on gonad differentiation in various amphibian species. The early gonad is bipotential, and in mammals, the expression of genes on the Y-chromosome is needed to masculinize the undifferentiated gonad; when this does not occur, the gonad develops into ovarian tissue. In Xenopus laevis frogs (and some other animals like birds), the opposite is true: females are heterogametic (i.e. ZW-chromosomes) and males have two of the same chromosomes (i.e. ZZ). In X. laevis, the W-chromosome is the dominant one, containing a gene, DM-W, which induces aromatase expression (331). Thus, having a W-chromosome is needed to produce estrogen; without the conversion of testosterone to estrogen, the frog develops as a male (332). Changes in sex ratio and gonadal morphology are therefore good indicators that an estrogen, or a chemical that up-regulates aromatase and indirectly increases estrogen levels, is present (76).
Determining a low-dose cutoff for atrazine is not a simple task. Although the safe limit of 3 μg/liter in drinking water was set by the EPA, actual levels in the environment often exceed this concentration (333), and levels in ponds and streams can reach 100 μg/liter (322) or more. In traditional toxicology studies examining several amphibian species, the LOAEL was set at 1.1 mg/liter, and the no observed effect level (NOEL) was 200 μg/liter (334, 335). Thus, using the definitions of low dose established by the NTP (2), we consider any treatment at or below 200 μg/liter to be a low dose.
In 2002, one of the first published studies to connect atrazine exposures to altered gonadal morphology examined X. laevis frogs exposed to 0.01–200 μg/liter throughout larval development (336). All doses from 0.1–200 μg/liter produced gonadal malformations including the presence of multiple gonads and hermaphroditism. Several other reports showed similar effects of low doses on gonadal phenotypes including studies that report the production of hermaphrodites and intersex frogs, males with ovotestes, and males with testicular oocytes (337–343). Additional studies showed that low-dose atrazine exposure (0.1–200 μg/liter in the water) during sexual differentiation caused testicular dysgenesis, testicular resorption, and testicular aplasia in male frogs (343, 344), and others indicated effects on sex ratios (339, 342, 345, 346). Importantly, these effects were not all observed at the same atrazine concentration, and the studies were conducted in several different species, with some reporting effects at low doses but no effects at higher doses (341) and others reporting effects in some but not all species (339). Examining these studies as a whole, there is clearly a pattern of effects that are reproducible from study to study, and they collectively support the hypothesis that atrazine disrupts sex hormone concentrations.
To date, five peer-reviewed studies have reported no effects of atrazine on sex ratios, gonadal morphology, the incidence of testicular abnormalities or testicular oocytes, gonad size, or the incidence of intersex phenotypes (347–351). Little can be ascertained from these negative studies, however, because four did not include any positive control, suggesting that the frogs used in those studies may have been incapable of responding to atrazine or any other hormonal treatment (347–350). Additionally, one of those studies reported testicular oocytes in the control frogs, suggesting either that the negative control population was contaminated with atrazine (or another EDC or hormone), or that an inappropriate strain of X. laevis was selected for the experiments (347). Only one study remains that did not find any effects of atrazine; this study used an appropriate positive control (17β-estradiol) and found effects of that hormone on sex ratios and the incidence of intersex gonads (351). An EPA expert panel noted, however, that this study used a strain of X. laevis that was obtained from a new, unexamined population of frogs from Chile and suggested that this strain may be insensitive to environmental chemicals. Furthermore, the panel called for additional analysis of the data in this study, including the statistical approaches; they suggested that an independent laboratory should evaluate the histopathological results; and they requested that atrazine metabolites be measured (352). The panel also proposed that these experiments should be repeated with an established X. laevis strain. Taking together the results of those studies that found effects of atrazine on sexual differentiation, and this one negative study, the WoE for the case of low-dose atrazine on sexual differentiation is clearly in support of adverse effects of this chemical.
Just as epidemiological studies have found links between EDCs and human diseases, ecological field studies have examined whether exposure to atrazine in natural environments affects the development of wild amphibians (343, 353–358). These studies have many of the same constraints as those observed in epidemiology: a paucity of data on early life exposures (including exposure levels of controls), limitations on the total number of EDCs that can be measured in environmental and biological samples, and a lack of causative relationships that can be established between exposures and effects. For these reasons, studies that found relationships between atrazine exposure (or concentrations in environmental samples) and effects on one or more aspect of sexual differentiation (343, 353–355) are considered weak, but significant, evidence for low-dose effects. The presence of several studies suggesting a relationship between low-dose exposure to atrazine in the wild and altered sexual differentiation indicates a plausible causal relationship. Because the ecological and laboratory data show similar effects of atrazine on gonadal development, this strengthens the conclusions of our WoE that low doses of atrazine cause harm to amphibians.
Feminization of males after atrazine exposure is not restricted to amphibians; exposure of zebrafish to low doses increased the ratio of female to male fish and increased expression of aromatase (359). Close to a dozen additional studies also report that environmentally relevant doses of atrazine can up-regulate aromatase, decrease testosterone, and/or increase estrogen levels in a large number of species (reviewed in Ref. 119), suggesting that low-dose effects of atrazine may be more widespread than their effects on the gonads of amphibians. Other studies indicate that low-dose atrazine affects the immune system and stress responses of salamanders (360–362), survivorship patterns of several frog species (363), and thyroid hormone and plasma ion concentrations in salmon (364).
An important factor to consider when examining the effects of atrazine on different animal models is the difficulty in identifying an appropriate low, environmentally relevant dose for all species. Aquatic animals can be housed in water containing levels of atrazine found in wild habitats, yet no toxicokinetic studies are available to determine what administered dose produces the levels of atrazine metabolites, typically in the parts-per-million or ppb range (365, 366), measured in human samples. There are also no blood or urine measurements in exposed rodents to compare with human levels; thus, extrapolations across species are estimates at best.
Keeping this qualification in mind, exposures in the range of 25–100 mg/kg · d during development have been shown to alter mammary gland development (367, 368), estrous cyclicity (369), serum and intratesticular testosterone concentrations (370), timing of puberty in males and prostate weight (371), and immune function (372) in rodents. Lower doses of atrazine metabolites (0.09–8.73 mg/kg · d) altered development of the mammary gland (373), male pubertal timing and prostate development (374). Identifying the range of doses administered to animals that produce the levels of atrazine and its metabolites measured in human blood and urine is an essential research need to pursue low-dose studies in rodents and other mammals.
F. Dioxin and spermatogenesis: low-dose effects from the most potent endocrine disruptor?
Dioxin, or TCDD, is formed as a byproduct of industrial processes as well as during waste incineration. Because TCDD is extremely toxic to some animals, with 1 μg/kg capable of killing 50% of guinea pigs, it has been labeled the most toxic chemical on earth (375). But interestingly, other animals are less sensitive to lethal effects of TCDD, with an LD50 of approximately 1000 μg/kg in hamsters, and studies also suggest that humans are not a hypersensitive species for lethality (376). Additionally, there are differences in the half-life of TCDD in different animals; in rodents, the half-life is 2–4 wks, but in humans, the half-life is approximately 10 yrs, and additional factors influence TCDD pharmacokinetics including the exposure level and the amount of body fat present (377–379). In cell cultures, doses as low as 10−11 m are toxic, with decreased viability observed even in cells maintained in nonproliferative states (380).
TCDD binds to the aryl hydrocarbon receptor (AhR), and differences in the affinity for the receptor may be responsible for differences in sensitivity between species (381). The Kd (dissociation constant for receptor-ligand binding kinetics) in human samples typically ranges from 3–15 nm, but in samples from rodents, the Kd is less than 1 nm (382). Importantly, there are also nongenomic pathways affected by TCDD that are mediated by AhR that are typically altered within minutes of TCDD exposure and therefore without changes in transcription (383). Yet many studies suggest that important differences exist between species regarding binding affinity of TCDD for AhR and the toxicity of this chemical, but that other adverse effects, including those related to the endocrine-disrupting activities of TCDD, occur at similar doses (or body burdens) across animal species (384, 385). Thus, it is plausible that AhR affinity alone can predict some, but not all, effects of TCDD and related chemicals.
The mechanisms responsible for many of the endocrine-disrupting activities of TCDD are currently not well understood. Knocking out AhR disrupts morphogenesis of several organ systems even in the absence of a ligand like TCDD, suggesting that this receptor plays important roles in early development (386). AhR is translocated to the nucleus after loss of cell-cell contacts and is often localized to the nucleus in embryonic cells, suggesting that it could have ligand-independent effects on development and/or that endogenous ligands could be present during early development (387). When TCDD is present, AhR translocates to the nucleus and dimerizes with ARNT, the aromatic hydrocarbon receptor nuclear translocator (388). Although the (currently unidentified) physiological activators of AhR are likely to induce rapid on/off signaling via AhR, TCDD and related compounds appear to maintain activation of AhR, and the presence of TCDD prevents the normal action of the AhR signaling pathway in the maintenance of homeostasis (389). This induces changes in the expression of genes and promotes the production of toxic metabolites. These effects may be responsible for some of the endocrine-related endpoints affected by TCDD exposure. Additionally, recent studies have shown complex and intricate interactions between the AhR and ER signaling pathways (390), suggesting that dioxin may also have indirect effects on some ER-mediated endpoints via AhR signaling.
Teratogenic effects of TCDD have been well documented after high-dose (391, 392) and low-dose exposures (393). These studies show that almost every organ and system in the body is affected by this chemical. High doses that did not produce lethality caused severe weight loss, intestinal hemorrhaging, alopecia, chloracne, edemas, and severe liver damage. Sadly, there are now several examples in humans of accidental exposures after the industrial release of TCDD where a number of individuals have been exposed to large doses (389, 394) as well as a few documented intentional poisonings (395). The tolerated daily intake level was set at 1–4 pg/kg · d, although the doses consumed by nursing infants are likely to exceed these levels by a factor of 10 (375). Adult exposures usually result from the consumption of contaminated foods, and because TCDD is lipophilic, it is concentrated in the fat component of breast milk and therefore passed in large quantities from a nursing mother to her infant.
Using classical toxicology methods, the effects of single TCDD doses were examined in adult male rats, specifically focusing on the effects of this chemical on the number of spermatids per testis and the integrity of the testicular germinal epithelium (396). In one of the earliest studies, Chahoud and colleagues (397) determined a LOAEL of 3 μg/kg · d and set the NOAEL at 1 μg/kg · d for effects on the testes. Because there are significant differences in the toxicity of TCDD between animal models, and different endpoints have different identified NOAELs, we have selected the 1 μg/kg · d identified by Chahoud et al. as the cutoff for low-dose studies of this compound. This cutoff is based on the NTP’s definition of low dose as occurring at doses lower than those tested in traditional toxicology assessments (2). However, it is important to acknowledge that body burdens that mimic those observed in human populations are likely the best indicators of low doses for TCDD (384), and thus we recommend that future studies determine body burdens after administration of TCDD for the specific strain, origin, and species of animal being tested to ensure that truly low doses, relevant to human populations, are being tested.
Several recent epidemiological studies have indicated that relatively high exposures to TCDD during early life (due to industrial release of high amounts of the chemical) can permanently affect semen quality and sperm count in men (398). Yet epidemiology studies also clearly show that the timing of TCDD exposure can vastly influence the effect of this chemical on spermatogenesis; exposures during perinatal life significantly reduced sperm parameters, but exposures during puberty increased sperm counts; exposures in adulthood had no effect on sperm parameters (399). Thus, it is also important for animal studies to focus on exposures during critical periods for development of the male reproductive tract and spermatogenesis in particular.
We are aware of 18 studies that have examined the effects of low doses (≤1 μg/kg · d) of TCDD during perinatal development on male fertility endpoints in adulthood. The endpoints assessed vary, including epididymal sperm counts, ejaculated sperm number, daily sperm production, sperm transit rate, and percent abnormal sperm, and the sensitivity of these endpoints appears to impact the ability to detect low-dose effects in different studies (400, 401) (Table 5). In total, 16 rodent studies examined the effect of low-dose TCDD on epididymal sperm count; 12 showed significant effects on this endpoint (402–413), whereas the other four did not (414–417). Of the five studies that examined ejaculated sperm counts, four studies (404, 405, 408), including one examining rhesus monkeys (418), showed effects of low-dose TCDD, i.e. a significant decrease in sperm counts; one study found no effect (417). Daily sperm production was a less-sensitive endpoint, with four studies showing significant decreases after prenatal exposure to low doses (402, 403, 407, 409) and four studies showing no effects (406, 412, 413, 416); sperm transit rate was examined in only two studies, although both showed significant decreases in sperm tranfer rates (403, 410); and finally, three studies determined that low-dose TCDD produced abnormalities in sperm appearance or motility (414, 415, 419), but one study was not able to replicate these findings (417).
Table 5.
Summary of low-dose animal studies examining the effects of TCDD on spermatogenesis endpoints
| Study | Administered dose (time of administration) | Animal | Epididymal sperm count | Ejaculated sperm no. | Daily sperm production | Sperm transit rate | % abnormal sperm |
|---|---|---|---|---|---|---|---|
| Mably et al. (409) | 0.064–1 μg/kg (gestational d 15) | Rat | Decreased | NA | Decreased | NA | NA |
| Bjerke and Peterson (402) | 1 μg/kg (gestational d 15) | Rat | Decreased | NA | Decreased | NA | NA |
| Gray et al. (404) | 1 μg/kg (gestational d 8) | Rat | Not significant | Decreased | NA | NA | NA |
| 1 μg/kg (gestational d 15) | Rat | Decreased | Decreased | NA | NA | NA | |
| 1 μg/kg (gestational d 11) | Hamster | Decreased | Decreased | NA | NA | NA | |
| Sommer et al. (408) | 1 μg/kg (gestational d 15) | Rat | Decreased | Decreased | Decreased | Not significant | Not significant |
| Wilker et al. (410) | 0.5, 1 or 2 μg/kg (gestational d 15) | Rat | Decreased | NA | Unaffected | Increased | NA |
| Gray et al. (405) | 0.05–1 μg/kg (gestational d 15) | Rat | Decreased | Decreased | Decreased | NA | NA |
| Faqi et al. (403) | 0.025–0.3 μg/kg (before mating, then 0.005–0.06 μg/kg weekly [to dams]) | Rat | Decreased | NA | Decreased | Increased | Increased |
| Loeffler and Peterson (412) | 0.25 μg/kg (gestational d 15) | Rat | Decreased | NA | Unaffected | NA | NA |
| Ohsako et al. (416) | 0.0125–0.8 μg/kg (gestational d 15) | Rat | Not significant | NA | Unaffected | NA | NA |
| Ohsako et al. (406) | 1 μg/kg (gestational d 15) | Rat | Decreased | NA | Unaffected | NA | NA |
| 1 μg/kg (gestational d 18) | Rat | Unaffected | NA | Unaffected | NA | NA | |
| 1 μg/kg (postnatal d 2 [to pups]) | Rat | Unaffected | NA | Unaffected | NA | NA | |
| Simanainen et al. (407) | 0.03–1 μg/kg (gestational d 15) | Rat | Decreased | NA | Decreased | NA | NA |
| Yonemoto et al. (417) | 0.0125–0.8 μg/kg (gestational d 15) | Rat | Unaffected | Unaffected | NA | NA | Unaffected |
| Yamano et al. (714) | 0.3 or 1 μg/kg (postnatal d 1 and then every week [to dams]) | Rat | Not significant | NA | NA | NA | NA |
| Ikeda et al. (715) | 0.4 μg/kg (before mating, then 0.08 μg/kg weekly [to dams]) | Rat | Unaffected | NA | NA | NA | NA |
| Bell et al. (414) | 0.05–1 μg/kg (gestational d 15) | Rat | Increased (at certain ages) | NA | NA | NA | Increased |
| Bell et al. (415) | 0.0024–0.046 μg/kg (d 12 weeks before pregnancy through parturition) | Rat | Unaffected | NA | NA | NA | Increased |
| Arima et al. (418) | 0.03 or 0.3 μg/kg (gestational d 20, then 5% of dose monthly [to dams]) | Rhesus monkey | Decreased | Not significant | NA | NA | Not significant |
| Yamano et al. (419) | 0.3 or 1 μg/kg (weekly to dams then pups [all postnatal]) | Rat | NA | NA | NA | NA | Increased |
| Jin et al. (411) | 1 μg/kg · d (postnatal days 1–4 [to dams]) | Mouse | Decreased | NA | NA | NA | NA |
| Rebourcet et al. (413) | 0.01–0.2 μg/kg (gestational d 15) | Rat | Decreased (at some ages) | NA | Not significant | NA | NA |
Not significant indicates trend for effect but did not reach statistical significance. Unaffected means assessed, but no differences were observed relative to controls. Here, low doses were considered any at or below 1 μg/kg · d (see text for discussion of how this cutoff was established for rodent studies). NA, Not assessed.
When examining the TCDD literature as a whole, the WoE strongly suggests that prenatal exposure to low doses of TCDD affects sperm-related endpoints in adulthood (Table 5). In all, only two studies were unable to detect any effect of TCDD on the sperm endpoints assessed, although both studies found effects of TCDD on other endpoints including the weight of the adult prostate (416) and the timing of puberty (417). No study on TCDD used a positive control, likely due to a paucity of information on the mechanisms of dioxin action, but this raises obvious questions about the ability of these experimental systems to detect effects on spermatogenesis. Finally, some of the inability to detect effects of TCDD could be due to the use of insensitive strains, because 1000-fold differences in sensitivity have been reported for different rodent strains (420).
Even though we have focused the majority of our attention on the effects of low-dose TCDD exposure on spermatogenesis, it should be noted that low doses of this chemical affect a multitude of endpoints in animals, altering immune function (421, 422), indicators of oxidative stress (423–425), bone and tooth development (426, 427), female reproduction and timing of puberty (428–430), mammary gland development and suceptibility to cancers (431), behaviors (432, 433), and others. In several cases, lower doses were more effective at altering these endpoints than higher ones (423, 424, 426, 433). Epidemiology studies of nonoccupationally exposed individuals also indicate that serum TCDD levels may be linked to diseases in humans as well (434). Mean serum TCDD levels have decreased by a factor of 7 over a 25-yr period (1972–97) in several industrial nations (435), but results from both animal and epidemiological studies suggest that even the low levels detected now could have adverse effects on health-related endpoints.
G. Perchlorate and thyroid: low-dose effects in humans?
A significant challenge with observing low-dose effects of EDCs in the human population is that human chemical exposures are multivariate along the vectors of time, space, and sensitivities. In addition, chemicals can exert effects on several systems simultaneously. Therefore, associations in human studies between exposures and disease are difficult to reconcile with experimental studies in animal model systems. For this reason, the literature describing the potential impacts of perchlorate contamination on the human population is potentially clarifying because to the best of our knowledge, perchlorate exerts only a single effect, and the pharmacology of perchlorate exposures has been studied in human volunteers (436). This literature offers a unique perspective into the issue of low-dose effects, perhaps providing important hypotheses to explain mechanistically why high-dose, short-term experiments can fail to predict the outcome of low-dose, lifetime exposures.
In the 2001–2002 NHANES dataset, perchlorate was detected in the urine of each of the 2820 samples tested (437). This widespread exposure means that the human population is being continuously exposed because perchlorate has a half-life in the human body of about 8 h (438). Human exposures to perchlorate are likely attributed to both contaminated drinking water and food (439); in fact, a recent analysis concludes that the majority of human exposure to perchlorate comes from food (440).
The predominant theory proposed to explain the source of perchlorate contamination in the United States is that it has been employed for many decades as the principal oxidant in explosives and solid rocket fuels (441). Perchlorate is chemically stable when wet and persists for long periods in geological systems and in ground water. Because of disposal practices during the 1960s through 1990s, perchlorate became a common contaminant of ground water in the United States (441, 442). Perchlorate is also formed under certain kinds of natural conditions (443), although the relative contributions to human exposure of these different sources is not completely understood. As a result of perchlorate contamination of natural waters, the food supply has become contaminated through irrigation in part because both aquatic and terrestrial plants can concentrate perchlorate more than 100-fold over water levels (444).
This exposure profile in the human population is important because high doses of perchlorate are known to reduce functioning of the thyroid gland, and poor thyroid function is an important cause of developmental deficits and adult disease (445). The primary question is: at what dose does perchlorate inhibit thyroid function sufficiently to cause disease? The current literature, reviewed below, supports the view that background exposure may affect thyroid function in adult women. These exposure levels, however, are considerably lower than predicted by early toxicology experiments in humans.
Perchlorate reduces thyroid function by inhibiting iodide uptake by the sodium/iodide symporter (NIS) (446), which is the only known effect of perchlorate on human physiology (438). NIS is responsible for transporting iodide into the thyroid gland, which is required for the production of thyroid hormone (447). However, NIS is also expressed in the gut (448, 449), in lactating breast (448, 450, 451), and in placenta (452), presumably all as a delivery mechanism for iodide to the developing and adult thyroid gland. Because the NIS transports perchlorate (450), the pathway by which humans take up and concentrate perchlorate is the same as the pathway by which humans take up and concentrate iodide. Interestingly, NIS expression in the human fetal thyroid gland is the rate-limiting step in production of thyroid hormone (453). Moreover, NIS transport of perchlorate explains why high levels of perchlorate are found in human amniotic fluid (454, 455) and breast milk (456–459).
This effect of perchlorate on thyroid function is important because thyroid hormone is essential for normal brain development, body growth as well as for adult physiology (445, 460). Moreover, it has become clear that even small deficits in circulating thyroid hormone in pregnant women (461, 462) or neonates (463) have permanent adverse outcomes. In fact, recent work indicates that very subtle thyroid hormone insufficiency in pregnant women is associated with cognitive deficits in their children (461). Because of the importance of thyroid hormone in development and adult physiology, and because perchlorate is a potent inhibitor of iodide uptake and thyroid hormone synthesis, identifying the dose at which these events occur is critical.
Perchlorate was used medically to reduce circulating levels of thyroid hormone in patients with an overactive thyroid gland in the 1950s and 1960s (reviewed in Ref. 446); therefore, it was reasonable to examine the dose-response characteristics of perchlorate on the human thyroid gland. Because perchlorate inhibits iodide uptake, several studies were performed to evaluate the effect of perchlorate exposure on iodide uptake inhibition in human volunteers (438, 464–466). In one study, 0.5 or 3 mg/d (approximately 0.007 and 0.04 mg/kg · d) perchlorate was administered to healthy volunteers (n = 9 females and 5 males, age 25–65 yr), and no effects were observed (466). Of course, it is important to note that the 2 wk of administration tested in this study is not sufficient to see any effect on serum concentrations of T4 or TSH; the healthy thyroid can store several months’ worth of thyroid hormone in the gland (467). Another small study also found no effects of administering 3 mg/d (approximately 0.04 mg/kg · d) on any thyroid endpoint assessed (n = 8 adult males) (464).
In contrast, two studies examining adult volunteers administered perchlorate found effects of this chemical on at least one endpoint. The first found that radioactive iodide uptake was affected by 2 wk of exposure to 10 mg/d (0.13 mg/kg · d), but other measures of thyroid function were not altered (n = 10 males) (465). The second examined adults (n = 37) given doses ranging from 0.007–0.5 mg/kg · d; all but the lowest dose altered radioactive iodide uptake, and only the highest dose altered TSH levels (438). These studies were interpreted to suggest that adults would have to consume 2 liters of drinking water daily that was contaminated with at least 200 ppb (200 μg/liter) perchlorate to reach a level in which iodide uptake would begin to be inhibited. Yet, these administered doses are high and relatively acute, so the derivation of a safe dose from these studies, applied to vulnerable populations such as those with low iodide intake, has been strongly disputed (471).
Studies of occupational exposures have also been used to examine the effects of exposure to relatively high levels of perchlorate. In the first such study, more than 130 employees were separated into eight groups based on exposure estimates from airborne perchlorate in the workplace (472). The authors found that individuals with longer daily exposures to perchlorate, due to longer work shifts, had significant decreases in TSH levels compared with individuals with shorter exposures. But this study was hampered because actual exposure levels were not measured via urine or blood samples. A second study examined 37 employees exposed to perchlorate and 21 control employees from an azide factory; actual exposure measures were not conducted, but estimates were calculated based on exposures to perchlorate dust and air samples (473). This study found no effects of perchlorate exposures on any thyroid endpoint, although the sample size examined was small. In the final occupational exposure study, serum perchlorate levels were measured and compared with several measures of thyroid function in workers (n = 29) who had spent several years as employees in a perchlorate production plant (474). In this study, the most complete because of the biomonitoring aspect of the exposure measures, higher perchlorate levels were associated with lower radioactive iodide uptake, higher urinary iodide excretion, and higher thyroid hormone concentrations.
Although iodide uptake was often inhibited in these studies, serum thyroid hormones were typically not altered, perhaps because of sufficient stored hormone. Based on these observations, the National Academy Committee to Assess the Health Implications of Perchlorate Ingestion (467) estimated that perchlorate would have to inhibit thyroid iodide uptake by about 75% for several months to cause a reduction in serum thyroid hormones. Moreover, the drinking water concentration of perchlorate required for this kind of inhibition was estimated to be over 1,000 ppb (438). Therefore, the National Academy of Sciences committee recommended a reference dose of 0.0007 mg/kg · d (467), based on the dose at which perchlorate could inhibit iodide uptake, and the EPA used this value to set a provisional drinking water standard of 15 ppb.
Considering these data and general knowledge about the thyroid system, it was unexpected that Blount et al. (475) would identify a positive association between urinary iodide and serum TSH in adult women in the NHANES 2001–2002 dataset. Yet several features of this dataset were consistent with a causal action of perchlorate on thyroid function. First, in the general population of adult women, urinary perchlorate was positively associated with serum TSH. In the population of adult women who also had low urinary iodide, however, urinary perchlorate was more strongly associated with serum TSH and was negatively associated with serum T4. The strength of this association was such that the authors calculated that women at the 50th percentile of perchlorate exposure experienced a 1 μg/dl T4 reduction (reference range = 5–12 μg/dl). Should this magnitude of reduction in serum T4 occur in a neonate, measurable cognitive deficits would also be present (476). Finally, Steinmaus et al. (477), using the same NHANES dataset, showed that women with low urinary iodide who smoke had an even stronger association between urinary perchlorate and measures of thyroid function. Tobacco smoke delivers thiocyanates, which also inhibit NIS-mediated iodide uptake (446).
The NHANES dataset suggests that perchlorate exposures of 0.2–0.4 μg/kg · d (440) are associated with depressed thyroid function, even when urinary iodide is not reduced. This is a considerably lower dose than the 7 μg/kg · d dose required to suppress iodide uptake in the Greer et al. (438) study or the 500 μg/kg · d the NAS estimated would be required for several months to actually cause a decline in serum T4. Therefore, it is reasonable to question whether these associations represent a causative relationship between perchlorate and thyroid function.
A number of epidemiological studies have been published to test for a relationship between perchlorate exposure and thyroid function. Early work used neonatal screening data for T4 as a measure of thyroid function, and the city of birth (Las Vegas, NV, compared with Reno, NV) as a proxy measure of exposure (478, 479). The reported findings were negative, but we now know that all Americans are exposed to perchlorate, so there was considerable misclassification of exposure, and no relationship should have been observed. Several additional studies using similar flawed designs also found no relationship between proxy measures of perchlorate exposures and clinical outcomes (480–484).
A recent study of the neonatal screening data from 1998 in California identified a strong association between neonatal TSH and whether or not the mother resided in a contaminated area (485). This study included over 497,000 TSH measurements and 800 perchlorate measurements. In addition, they used as a cutoff a variety of TSH levels (as opposed to the 99.9th percentile used for the diagnosis of congenital hypothyroidism), indicating that perchlorate exposure is not associated with congenital hypothyroidism. Two additional studies have shown similar relationships between perchlorate and TSH levels, particularly in families with a history of thyroid disease (486, 487).
Several studies in pregnant women have failed to identify a relationship between perchlorate exposure and measures of thyroid function (488–490). Although these are important studies that need to be carefully scrutinized, they do not replicate or refute the NHANES dataset. It thus remains important to conduct additional studies exploring the relationship between background exposure to perchlorate and thyroid function in adults, pregnant women, neonates, and infants. This effort will be challenging because of the different characteristics of thyroid function and hormone action at different life stages (460). In addition, it will be important to obtain individual measurements of exposures to perchlorate and other NIS inhibitors (thiocyanate and nitrate), and iodide itself as well as individual measures of thyroid function (free and total T4 and TSH).
If background levels of perchlorate affect thyroid function in any segment of the population, it will be challenging to explain how the high-dose, short-term experiments of Greer et al. (438) completely underestimated the sensitivity of the human thyroid gland to perchlorate exposure. One possibility is that physiological systems respond to short durations of robust stress with compensatory mechanisms that reset during periods of long-term stress.
When these data are examined together, several important issues are raised. First, this example illustrates the difficulties inherent in studying human populations; epidemiology yields associations, not cause-effect relationships, in many cases using surrogate markers for perchlorate, and is not able to distinguish short- vs. long-term exposure duration. Second, our WoE analysis suggests that there is weak evidence for low-dose effects of perchlorate; further research is needed. The relationship between low-dose perchlorate exposures and thyroid endpoints would be strengthened by the addition of studies that measure biological concentrations of perchlorate and compare them with thyroid endpoints in neonates and other vulnerable populations. Third, the published studies that reported low-dose effects of perchlorate typically examined very specific populations, with several focusing on women with low iodine intake. This observation suggests that some groups may be more vulnerable to low doses of perchlorate than others (491).
H. Low-dose summary
These examples, and the examples of low-dose effects in less well-studied chemicals (Table 3), provide evidence that low-dose effects are common in EDC research and may be the default expectation for all chemicals with endocrine activity. Many known EDCs have not been examined for low-dose effects, but we predict that these chemicals will have effects at low doses if studied appropriately. Although studies unable to detect effects at low doses have received attention, including some studies designed to replicate others that reported low-dose effects, the majority of these studies contain at least one major design flaw. Thus, a WoE approach clearly indicates that low-dose effects are present across a wide span of chemical classes and activities.Go to:
III. Nonmonotonicity in EDC Studies
A concept related to low dose is that of nonmonotonicity. As noted in Section I.B, in a monotonic response, the observed effects may be linear or nonlinear, but the slope does not change sign (Fig. 3, A and B). In contrast, a dose-response curve is nonmonotonic when the slope of the curve changes sign somewhere within the range of doses examined (Fig. 3C). NMDRCs are often U-shaped (with maximal responses of the measured endpoint observed at low and high doses) or inverted U-shaped (with maximal responses observed at intermediate doses) (Fig. 3C, top panels). Some cases are more complicated, with multiple points along the curve at which the slope of the curve reverses sign (Fig. 3C, bottom left). Nonmonotonicity is not synomymous with low dose, because there are low-dose effects that follow monotonic dose-response curves. Thus, it is not required that a study include doses that span from the true low-dose range to the high toxicological range to detect nonmonotonicity. The consequence of NMDRCs for toxicity testing is that a safe dose determined from high doses does not guarantee safety at lower, untested doses that may be closer to current human exposures.

Examples of NMDRCs from the cell culture, animal, and epidemiological literature will be discussed in detail in Section III.C. Importantly, our review of the literature finds that NMDRCs are common in the endocrine and EDC literature. In fact, it is plausible that, considering the mechanisms discussed below, NMDRCs are not the exception but should be expected and perhaps even common.
A. Why is nonmonotonicity important?
NMDRCs in toxicology and in the regulatory process for EDCs are considered controversial. In addition to discussions of whether NMDRCs exist, there is also discussion of whether those that do exist have relevance to toxicological determination of putative safe exposures. In the standard practice of regulatory toxicology, the calculated safe dose, also called a reference dose, is rarely tested. In a system that is responding nonmonotonically, it is not appropriate to use a high-dose test to predict low-dose effects. Unfortunately, all regulatory testing for the effects of chemical exposures assume that this is possible. All current exposure standards employed by government agencies around the world, including the FDA and EPA, have been developed using an assumption of monotonicity (492, 493). The low-dose range, which presumably is what the general public normally experiences, is rarely, if ever, tested directly.

As described above, there are other nonlinear dose-response curves that are monotonic (Fig. 3B). These curves may also present problems for extrapolating from high doses to low doses because there is no linear relationship that can be used to predict the effects of low doses. Equally troubling for regulatory purposes are responses that have a binary response rather than a classical dose-response curve (Fig. 3D). In these types of responses, one range of doses has no effect on an endpoint, and then a threshold is met, and all higher doses have the same effect. An example is seen in the atrazine literature, where doses below 1 ppb had no effect on the size of the male larynx but doses at or above 1 ppb produced a significant decrease in size of approximately 10–15% (336). Even doses of 200 ppb, the toxicological NOEL, produce the same effect. Thus, this all-or-none effect is observed because atrazine does not shrink the larynx; instead, it removes the stimulatory agent (i.e. androgens). In the absence of some threshold dose of androgen, the larynx simply remains at the unstimulated (female) size. The EPA’s assessment of this study and others was that the lack of a dose-dependent response negates the importance of this effect (352). The lack of a dose response for a threshold effect like larynx size does not mean that the effects are not dose dependent; thus, understanding these types of effects and their implications for risk assessments is essential for determining the safe levels of chemicals.
It is important to mention here that the appropriateness of determining NOAEL concentrations, and therefore calculating reference doses, from exposures to endogenous hormones or EDCs has been challenged by several studies (Fig. 4A) (494–496). These studies show that hormonally active agents may still induce significant biological effects even at extremely low concentrations and that presently available analytical methods or technologies might be unable to detect relatively small magnitudes of effects. Previous discussions of this topic have shown that as the dose gets lower (and approaches zero) and the effect size decreases, the number of animals needed to achieve the power to detect a significant effect would have to increase substantially (497). Even more importantly, the assumption of a threshold does not take into account situations where an endogenous hormone is already above the dose that causes detectable effects and that an exogenous chemical (whether an agonist or antagonist) will modulate the effect of the endogenous hormone at any dose above zero (Fig. 4B). There can thus be no threshold or safe dose for an exogenous chemical in this situation. Forced identification of NOAEL or threshold doses based on the assumption that dose-response curves are always monotonic without considering the background activity of endogenous hormones and the limitations of analytical techniques supports the misconception that hormonally active agents do not have any significant biological effects at low doses. Thus, the concept that a toxic agent has a safe dose that can be readily estimated from the NOAEL derived from testing high, acutely toxic doses is overly simplistic and contradicted by data when applied to EDC (5, 497, 498).
B. Mechanisms for NMDRCs
Previously, the lack of mechanisms to explain the appearance of NMDRCs was used as a rationale for ignoring these phenomena (492, 493). This is no longer acceptable because there are several mechanisms that have been identified and studied that demonstrate how hormones and EDCs produce nonmonotonic responses in cells, tissues, and animals. These mechanisms include cytotoxicity, cell- and tissue-specific receptors and cofactors, receptor selectivity, receptor down-regulation and desensitization, receptor competition, and endocrine negative feedback loops. These mechanisms are well understood, and by providing detailed biological insights at the molecular level into the etiology of NMDRCs, they strongly negate the presumption that has been central to regulatory toxicology that dose-response curves are by default monotonic.
1.Cytotoxicity
The simplest mechanism for NMDRCs derives from the observation that hormones can be acutely toxic at high doses yet alter biological endpoints at low, physiologically relevant doses. Experiments working at concentrations that are cytotoxic are incapable of detecting responses that are mediated by ligand-binding interactions. For example, the MCF7 breast cancer cell line proliferates in response to estradiol in the low-dose range (10−12 to 10−11 m) and in the pharmacological and toxicological range (10−11 to 10−6 m), but toxic responses are observed at higher doses (38). Thus, when total cell number is graphed, it displays an inverted U-shaped response to estrogen. But cells that do not contain ER, and therefore cannot be affected by the hormonal action of estradiol, also display cytotoxic responses when treated with high doses of hormone. These results clearly indicate that the effects of estradiol at high doses are toxic via non-ER-mediated mechanisms.
2. Cell- and tissue-specific receptors and cofactors
Some NMDRCs are generated by the combination of two or more monotonic responses that overlap, affecting a common endpoint in opposite ways via different pathways. For example, in vitro cultured prostate cell lines demonstrate a nonmonotonic response to increasing doses of androgen where low doses increase cell number and higher doses decrease cell number, thus producing an inverted U-shaped curve (499, 500). Although the parental cell expressed an inverted U-shaped dose-response curve, after a long period of inhibition, the effects on cell number could be segregated by selecting two populations of cells: one that proliferated in the absence of androgens and other cells that proliferated in the presence of high androgen levels (501). Thus, the observed inverted U-shaped response is due to actions via two independent pathways that can be separated from each other in an experimental setting (502). Similarly, estrogens have been shown to induce cell proliferation and inhibit apoptosis in several cell populations, but inhibit proliferation and induce apoptosis in others (503, 504), with the combined effect being an inverted U-shaped curve for cell number (505).
Why does one single cell type have different responses to different doses of the same hormone? The case of the prostate cell line described above is reminiscent of the results described from the transcriptome of MCF7 cells, whereby a discrete global response like cell proliferation manifests at significantly lower estrogen doses than the induction of a single marker gene (135). That a response like cell proliferation requires a significantly lower dose of hormone than the dose needed to induce a given target gene is counterintuitive but factual; it may be interpreted as consistent with the notion that metazoan cells, like cells in unicellular organisms, are intrinsically poised to divide (503, 506, 507) and that quiescence is an induced state (508, 509). The biochemical details underlying these different responses are largely unknown; however, recent studies showed that steroid receptors control only a portion of their target genes directly via promoter binding. The majority of the changes are indirect, through chromatin rearrangements (510, 511).
Why do different cell types (in vitro and in vivo) have different responses to the same hormone? One answer is that they may express different receptors, and these receptors have different responses to the same hormone. For example, some tissues express only one of the two major ER (ERα and ERβ), and actions via these receptors are important not just for responsiveness to hormone but also for cellular differentiation and cross talk between tissue compartments (512). Yet other tissues express both ERα and ERβ, and the effects of signaling via these two receptors often oppose each other; i.e. estrogen action via ERα induces proliferation in the uterus, but ERβ induces apoptosis (154). Complicating the situation further, different responses to a hormone can also be obtained due to the presence of different cofactors in different cell and tissue types (513, 514); these coregulators influence which genes are transcriptionally activated or repressed in response to the presence of hormone. They can also influence ligand selectivity of the receptor and DNA-binding capacity, having tremendous impact on the ability of a hormone to have effects in different cell types (105, 515, 516).
Although much of these activities occur on a biochemical level, i.e. at the receptor, there is also evidence that nonmonotonicity can originate at the level of tissue organization. The mammary gland has been used as a model to study inter- and intracompartmental effects of hormone treatment: within the ductal epithelium, estrogen has distinct effects during puberty, both inducing proliferation, which causes growth of the ductal tree, and inducing apoptosis, which is required for lumen formation (517, 518); in cell culture, the presence of stromal cells can also enhance the effects of estrogen on epithelial cells (519, 520), suggesting that stromal-epithelial compartmental interactions can mediate the effects of estrogen.
3. Receptor selectivity
NMDRCs can occur because of differences in receptor affinity, and thus the selectivity of the response, at low vs. high doses. For example, at low doses, BPA almost exclusively binds to the ER (including mER), but at high doses it can also bind weakly to other hormone receptors, like androgen receptor and thyroid hormone receptor (249, 521). This type of receptor nonselectivity is quite common for EDCs, and it has been proposed that binding to different receptors may be an explanation for the diverse patterns of disease observed after EDC exposures (522). In fact, several of the chemicals shown to have low-dose effects are known to act via multiple receptors and pathways (Table 3). Thus, the effects seen at high doses can be due to action via the binding of multiple receptors, compared with the effects of low doses, which may be caused by action via only a single receptor or receptor family.
4. Receptor down-regulation and desensitization
When hormones bind to nuclear receptors, the ultimate outcome is a change in the transcription of target genes. When the receptor is bound by ligand, an increase in response is observed; as discussed previously in this review, the relationship between hormone concentration and the number of bound receptors, as well as the relationship between the number of bound receptors and the biological effect, is nonlinear (38). After the nuclear receptor is bound by hormone and transcription of target genes has occurred (either due to binding of the receptor at a DNA response element or the relief of a repressive event on the DNA), the reaction eventually must cease; i.e. the bound receptor must eventually be inactivated in some way. Thus, nuclear hormone receptors are ubiquitinated and degraded, usually via the proteasome (523). Importantly, the role of the hormone in receptor degradation differs depending on the hormone; binding of estrogen, progesterone, and glucocorticoid mediates the degradation of their receptors (524–526), whereas the presence of hormone may actually stabilize some receptors and prevent degradation (527), and other receptors are degraded without ligand (528). As hormone levels rise, the number of receptors being inactivated and degraded also rises, and eventually the number of receptors being produced cannot maintain the pace of this degradation pathway (523). Furthermore, the internalization and degradation of receptors can also influence receptor production, leading to an even stronger down-regulation of receptor (529). In the animal, the role of receptor down-regulation is actually quite complex, because signaling from one hormone receptor can influence protein levels of another receptor; i.e. ER signaling can promote degradation of the glucocorticoid receptor by increasing the expression of enzymes in the proteasome pathway that degrade it (530).
There is also the issue of receptor desensitization, a process whereby a decrease in response to a hormone is not due to a decrease in the number of available receptors but instead due to the biochemical inactivation of a receptor (531). Desensitization typically occurs when repeated or continuous exposure to ligand occurs. Normally seen with membrane-bound G protein-coupled receptors, the activation of a receptor due to ligand binding is quickly followed by the uncoupling of the activated receptor from its G proteins due to phosphorylation of these binding partners (532). Receptor desensitization has been observed for a range of hormones including glucagon, FSH, human chorionic gonadotropin, and prostaglandins (533). Importantly, desensitization and down-regulation can occur in the same cells for the same receptor (534), and therefore, both can play a role in the production of NMDRCs.
5. Receptor competition
Mathematical modeling studies suggest that the mixture of endogenous hormones and EDCs establishes a natural environment to foster NMDRCs. Using mathematical models, Kohn and Melnick (42) proposed that when EDC exposures occur in the presence of endogenous hormone and unoccupied hormone receptors, some unoccupied receptors become bound with the EDC, leading to an increase in biological response (i.e. increased expression of a responsive gene, increased weight of an organ, etc.). At low concentrations, both the endogenous hormone and the EDC bind to receptors and activate this response, but at high doses, the EDC can outcompete the natural ligand. The model predicts that inverted U-shaped curves would occur regardless of the binding affinity of the EDC for the receptor and would be abolished only if the concentration of natural hormone were raised such that all receptors were bound.
6. Endocrine negative feedback loops
In several cases, the control of hormone synthesis is regulated by a series of positive- and negative feedback loops. Several hormones are known to control or influence their own secretion using these feedback systems. In one example, levels of insulin are known to regulate glucose uptake by cells. Blood glucose levels stimulate insulin production, and as insulin removes glucose from circulation, insulin levels decline. Thus, NMDRCs can occur as the free/available ligand and receptor concentrations are influenced by one another. In another example, thyroid hormone secretion is stimulated by TSH, and thyroid hormone suppresses TSH; thus, feedback between these two hormones allows thyroid hormone to be maintained in a narrow dose range.
Several studies indicate that these negative feedback loops could produce NMDRCs when the duration of hormone administration is changed (535). For example, short exposures of estrogen induce proliferation in the uterus and pituitary, but longer hormone regimens inhibit cell proliferation (236, 536). Thus, the outcome is one where exposure to a single hormone concentration stimulates an endpoint until negative feedback loops are induced and stimulation ends (537).
7. Other downstream mechanisms
Removing the variability that can come from examining different cell types, or even single cell types in the context of a tissue, studies of cultured cells indicate that different gene profiles are affected by low doses of hormone compared with higher doses. In a study of the genes affected by low vs. higher doses of estrogen, researchers found that there were a small number of genes in MCF7 breast cancer cells with very high sensitivity to low doses of estradiol (10 pm) compared with the total number of genes that were affected by higher (30 or 100 pm) exposures (538). But the surprising finding was the pattern of estradiol-induced vs. estradiol-suppressed gene expression at high and low doses; when 10 pm was administered, the number of estradiol-suppressible genes was approximately three times higher than the number of estradiol-inducible genes. However, the overall profile of the number of estradiol-suppressible genes was approximately half the total number of estradiol-inducible genes. This observation suggests that low doses of estrogen selectively target a small subset of the total number of estrogen-sensitive genes and that the genes affected by low doses are most likely to be suppressed by that treatment. The mechanisms describing how low doses of estrogen differently affect the expression of genes compared with higher doses have yet to be elucidated, but low doses of estradiol inhibit expression of apoptotic genes (539), indicating that which genes are affected by hormone exposure is relevant to understand how low doses influence cellular activities.
C. Examples of nonmonotonicity
1. Examples of NMDRCs from cell culture
A tremendous amount of theoretical and mathematical modeling has been conducted to understand the production of nonlinear and nonmonotonic responses (42, 540). These studies and others suggest that the total number of theoretical response curves is infinite. Yet this does not mean that the occurrence of NMDRCs is speculative; these types of responses are reported for a wide variety of chemicals. Cell culture experiments alone provide hundreds of examples of nonmonotonic responses (see Table 6 for examples). In the natural hormone category, many different hormones produce NMDRCs; this is clearly not a phenomenon that is solely attributable to estrogen and androgen, the hormones that have been afforded the most attention in the dose-response literature. Instead, NMDRCs are observed after cells are treated with a range of hormones, suggesting that this is a fundamental and general feature of hormones.
Table 6.
Examples of NMDRCs in cell culture experiments
| Chemicals by chemical class | Nonmonotonic effect | Cell type | Refs. |
|---|---|---|---|
| Natural hormones | |||
| 17β-Estradiol | Cell number | MCF7 breast cancer cells | 135, 716 |
| Dopamine uptake | Fetal hypothalamic cells (primary) | 717 | |
| pERK levels, prolactin release | GH3/B6/F10 pituitary cells | 41, 718, 719 | |
| β-Hexosaminidase release | HMC-1 mast cells | 720 | |
| Cell number | Vascular smooth muscle cells | 721 | |
| Production of l-PGDS, a sleep-promoting substance | U251 glioma cells | 722 | |
| 5α-Dihydrotestosterone | Cell number | LNCaP-FGC prostate cancer cells | 499 |
| Cell number, kinase activity | Vascular smooth muscle cells | 721 | |
| 5α-Androstenedione | Cell number | LNCaP-FGC prostate cancer cells | 499 |
| Corticosterone | Mitochrondrial oxidation, calcium flux | Cortical neurons (primary) | 723 |
| Insulin | Markers of apoptosis (in absence of glucose) | Pancreatic β-cells (primary) | 724 |
| Progesterone | Cell number | LNCaP-FGC prostate cancer cells | 499 |
| Prolactin | Testosterone release | Adult rat testicular cells (primary) | 725 |
| hCG | Testosterone release | Adult rat testicular cells (primary) | 725 |
| T3 | Rate of protein phosphorylation | Cerebral cortex cells (primary, synaptosomes) | 726 |
| LPL mRNA expression | White adipocytes (rat primary) | 727 | |
| GH | IGF-I expression | Hepatocytes (primary cultures from silver sea bream) | 728 |
| Pharmaceutical hormones | |||
| DES | Cell number | MCF7 breast cancer cells | 716 |
| Prolactin release | GH3/B6/F10 pituitary cells | 41 | |
| Ethinyl estradiol | CXCL12 secretion | MCF7 breast cancer cells, T47D breast cancer cells | 729 |
| R1881 (synthetic androgen) | Cell number | LNCaP-FGC cells | 499 |
| Trenbolone | Induction of micronuclei | RTL-W1 fish liver cells | 730 |
| Plastics | |||
| BPA | Cell number | MCF7 breast cancer cells | 135, 716 |
| Dopamine efflux | PC12 rat tumor cells | 40 | |
| pERK levels, intracellular Ca2+ changes, prolactin release | GH3/B6/F10 pituitary cells | 41, 718 | |
| Cell number | LNCaP prostate cancer cells | 731 | |
| DEHP | Number of colonies | Escherichia coli and B. subtilis bacteria | 732 |
| Di-n-octyl phthalate | Number of colonies | E. coli and B. subtilis bacteria | 732 |
| Detergents, surfactants | |||
| Octylphenol | Cell number | MCF7 breast cancer cells | 716 |
| Dopamine uptake | Fetal hypothalamic cells (primary) | 717 | |
| pERK levels | GH3/B6/F10 pituitary cells | 718 | |
| hCG-stimulated testosterone levels | Leydig cells (primary) | 733 | |
| Propylphenol | pERK levels | GH3/B6/F10 pituitary cells | 718 |
| Nonylphenol | pERK levels, prolactin release | GH3/B6/F10 pituitary cells | 41, 718 |
| β-Hexosaminidase release | HMC-1 mast cells | 720 | |
| Cell number | MCF7 breast cancer cells | 135 | |
| PAH | |||
| Phenanthrene | All-trans retinoic acid activity | P19 embryonic carcinoma cells | 734, 735 |
| Benz(a)acridine | All-trans retinoic acid activity | P19 embryonic carcinoma cells | 734 |
| Naphthalene | hCG-stimulated testosterone | Pieces of goldfish testes | 736 |
| Β-naphthoflavone | hCG-stimulated testosterone | Pieces of goldfish testes | 736 |
| Retene | hCG-stimulated testosterone | Pieces of goldfish testes | 736 |
| Heavy metals | |||
| Lead | Estrogen, testosterone, and cortisol levels | Postvitellogenic follicles (isolated from catfish) | 737 |
| Cadmium | Expression of angiogenesis genes | Human endometrial endothelial cells | 738 |
| Phytoestrogens and natural antioxidants | |||
| Genistein | Cell number | Caco-2BBe colon adenocarcinoma cells | 739 |
| CXCL12 secretion, cell number | T47D breast cancer cells | 729 | |
| Cell number, cell invasion, MMP-9 activity | PC3 prostate cancer cells | 740 | |
| pJNK levels, Ca2+ flux | GH3/B6/F10 pituitary cells | 719 | |
| Coumesterol | Prolactin release, pERK levels | GH3/B6/F10 pituitary cells | 719 |
| Daidezin | Prolactin release, pERK levels | GH3/B6/F10 pituitary cells | 719 |
| Cell number | MCF7 breast cancer cells | 135 | |
| Cell number | LoVo colon cancer cells | 741 | |
| Resveratrol | Expression of angiogenesis genes | Human umbilical vein endothelial cells | 742 |
| Trans-resveratrol | pERK levels, Ca2+ flux | GH3/B6/F10 pituitary cells | 719 |
| Artelastochromene | Cell number | MCF7 breast cancer cells | 743 |
| Carpelastofuran | Cell number | MCF7 breast cancer cells | 743 |
| Biochanin A | Induction of estrogen-sensitive genes in the presence of testosterone | MCF7 breast cancer cells | 744 |
| Licoflavone C | Induction of estrogen-sensitive genes | Yeast bioassay | 745 |
| Quercetin | Aromatase activity | H295R adrenocortical carcinoma cells | 746 |
| Cell number | SCC-25 oral squamous carcinoma cells | 747 | |
| Dioxin | |||
| TCDD | Cell number, gene expression | M13SV1 breast cells | 748 |
| PCB | |||
| PCB-74 | Cell viability, GnRH peptide levels | GT1-7 hypothalamic cells | 749 |
| PCB-118 | Cell viability, GnRH peptide levels | GT1-7 hypothalamic cells | 749 |
| Aroclor 1242 (PCB mixture) | β-Hexosaminidase release | HMC-1 mast cells | 720 |
| POP mixture | Apoptosis of cumulus cells | Oocyte-cumulus complexes (primary, isolated from pigs) | 750 |
| Herbicides | |||
| Glyphosphate-based herbicide (Round-Up) | Cell death, aromatase activity, ERβ activity | HepG2 liver cells | 751 |
| Atrazine | Cell number | IEC-6 intestinal cells | 752 |
| Insecticides | |||
| Endosulfan | Cell number | IEC-6 intestinal cells | 752 |
| β-Hexosaminidase release | HMC-1 mast cells | 720 | |
| ATPase activity of P-glycoprotein | CHO cell extracts | 753 | |
| Diazinon | Cell number | IEC-6 intestinal cells | 752 |
| Dieldrin | β-Hexosaminidase release | HMC-1 mast cells | 720 |
| DDT | Cell number | MCF7 breast cancer cells | 144 |
| DDE | β-Hexosaminidase release | HMC-1 mast cells | 720 |
| Prolactin release | GH3/B6/F10 pituitary cells | 41 | |
| 3-Methylsulfonyl-DDE | Cortisol and aldosterone release, expression of steroidogenic genes | H295R adrenocortical carcinoma cells | 754 |
| Fungicides | |||
| Hexachlorobenzene | Transcriptional activity in the presence of DHT | PC3 prostate cancer cells | 755 |
| Prochloraz | Aldosterone, progesterone, and corticosterone levels; expression of steroidogenic genes | H295R adrenocortical cells | 756 |
| Ketoconazole | Aldosterone secretion | H295R adrenocortical cells | 757 |
| Fungicide mixtures | Aldosterone secretion | H295R adrenocortical cells | 757 |
| PBDE | |||
| PBDE-49 | Activation of ryanodine receptor 1 | HEK293 cell (membranes) | 758 |
| PBDE-99 | Expression of GAP43 | Cerebral cortex cells (primary) | 759 |
Chemicals from a large number of categories with variable effects on the endocrine system also produce NMDRCs in cultured cells. These chemicals range from components of plastics to pesticides to industrial chemicals and even heavy metals. The mechanisms for nonmonotonicity discussed in Section III.B are likely explanations for the NMDRCs reported in a range of cell types after exposure to hormones and EDCs. Table 6 provides only a small number of examples from the literature, and it should be noted that because these are studies of cells in culture, most of these studies typically examined only a few types of outcomes: cell number (which could capture the effects of a chemical on cell proliferation, apoptosis, or both), stimulation or release of another hormone, and regulation of target protein function, often examined by measuring the phosphorylation status of a target.
2. Examples of NMDRCs in animal studies
Some scientists suggest that nonmonotonicity is an artifact of cell culture, however, a large number of NMDRCs have been observed in animals after administration of natural hormones and EDCs, refuting the hypothesis that this is a cell-based phenomenon only. Similar to what has been observed in cultured cells, the NMDRCs observed in animals also span a large range of chemicals, model organisms, and affected endpoints (Table 7). These results underscore the biological importance of the mechanisms of nonmonotonicity that have been largely worked out in vitro.
Table 7.
Examples of NMDRCs in animal studies
| Chemicals by chemical class | Nonmonotonic effect | Organ/sex/animal | Refs. |
|---|---|---|---|
| Natural hormones | |||
| 17β-Estradiol | Morphological parameters | Mammary gland/female/mice | 138, 541 |
| Accumulation of cAMP | Pineal/female/rats | 760 | |
| Prostate weight | male/mice | 689 | |
| Uterine weight | female/mice | 761 | |
| Antidepressant effects, measured by immobility assay | Behavior/male/mice | 762 | |
| Nocturnal activity, gene expression in preoptic area | Brain and behavior/female/mice | 763 | |
| Corticosterone | Spatial memory errors | Behavior/male/rats | 764 |
| Cholinergic fiber loss in cortex after treatment with neurodegenerative drugs | Brain/male/rats | 765 | |
| Mitochondrial metabolism | Muscle/male/rats: strain differences | 766 | |
| Contextual fear conditioning | Behavior/male/rats | 767 | |
| Locomotor activity | Behavior/male/captive Adelie penguins | 768 | |
| Glucocorticoid | Na+/K+-ATPase activity | Brain/tilapia (fish) | 769 |
| Testosterone | Na+/K+-ATPase activity | Brain/tilapia (fish) | 769 |
| Gonadotropin subunit gene expression | Pituitary/sexually immature goldfish | 770 | |
| 11β-Hydroxyandrosterone | Gonadotropin subunit gene expression | Pituitary/sexually immature goldfish | 770 |
| T4 | Bone growth | Tibia/male/rats with induced hypothyroidism | 771 |
| Leptin | Insulin production (in the presence of glucose) | Pancreas/male/rats | 560 |
| Oxytocin | Infarct size, plasma LDH levels, creatine kinase activity after ischemia/ reperfusion injury | Brain and blood/male/rats | 772 |
| Memory retention | Behavior/male/mice | 773 | |
| Melatonin | Brain infarction and surviving neuron number after injury | Brain/female/rats | 774 |
| Dopamine | Memory | Brain/both/rhesus monkey | 775 |
| Neuronal firing rate | Brain/male/rhesus monkey | 776 | |
| Pharmaceutical | |||
| DES | Sex ratio, neonatal body weight, other neonatal development | Mice | 777 |
| Adult prostate weight | Male/mice | 689 | |
| Uterine weight | Female/mice | 761 | |
| Expression of PDGF receptor | Testes/male/rats | 778 | |
| Morphological parameters | Mammary gland/male and female/mice | 779 | |
| Estradiol benzoate | Dorsal prostate weight, body weight | Male/rats | 780 |
| Sexual behaviors, testes morphology | Male/zebra finches (birds) | 781 | |
| Ethinyl estradiol | GnRH neurons | Brain/zebrafish | 782 |
| Tamoxifen | Uterine weight | Female/mice | 761 |
| Fluoxetine (antidepressant) | Embryo number | Potamopyrgus antipodarum (snails) | 783 |
| Fadrozole (aromatase inhibitor) | Aromatase activity | Ovary/female/fathead minnows | 784 |
| Plastics | |||
| BPA | Fertility | Reproductive axis /female/mice | 316 |
| Reproductive behaviors | Behavior/male/rats | 785 | |
| Protein expression | Hepatopancreas/male/Porcellio scaber (isopod) | 786 | |
| Timing of vaginal opening, tissue organization of uterus | Reproductive axis/female/mice | 577 | |
| Expression of receptors in embryos | Brain and gonad/both/ mice | 787 | |
| DEHP | Aromatase activity | Hypothalamus/male/rats | 788 |
| Cholesterol levels | Serum/male/rats | 569 | |
| Timing of puberty | Reproductive axis /male/rats | 789 | |
| Body weight at birth, vaginal opening, and first estrous | Female/rats | 790 | |
| Seminal vesicle weight, epididymal weight, testicular expression of steroidogenesis genes | Male/rats | 791 | |
| Responses to allergens, chemokine expression | Skin/male/mice | 792 | |
| Detergents, surfactants | |||
| Nonylphenol ethoxylate | Fecundity | Biomphalaria tenagophila (snails) | 793 |
| Octylphenol | Embryo production | P. antipodarum (snails) | 794 |
| Spawning mass and egg numbers | Marisa cornuarietis (snails) | 795 | |
| Semicarbazide | Timing of preputial separation, serum DHT | Male/rats | 796 |
| Antimicrobial | |||
| Triclocarban | Fecundity | P. antipodarum (snails) | 797 |
| PCB | |||
| Mixture of PCB | Corticosterone levels | Male/kestrels (birds) | 798 |
| Environmental PCB mixture | Corticosterone levels | Female/tree swallows (birds) | 799 |
| UV filters | |||
| Octyl methoxycinnamate | Activity, memory | Behavior/both/rats | 800 |
| Aromatic hydrocarbons | |||
| Β-naphthoflavone | Testosterone | Plasma/male/goldfish | 736 |
| Toluene | Locomotor activity | Behavior/male/rats | 801 |
| Dioxins | |||
| TCDD | Cell-mediated immunity | Immune system/male/ rats | 802 |
| Proliferation after treatment with chemical carcinogen | Liver/female/rats | 803 | |
| Heavy metals | |||
| Cadmium | Expression of metallothionein, pS2/TFF1 | Intestine and kidney/ female/rats | 804 |
| Activity of antioxidant enzymes | Earthworms | 805 | |
| Size parameters, metamorphic parameters | Xenopus laevis | 806 | |
| Lead | Growth, gene expression | Vicia faba seedlings (plant) | 807 |
| Retinal neurogenesis | Eye and brain/female/rats | 808 | |
| Selenium | DNA damage, apoptotic index | Prostate/male/dogs | 809 |
| Hatching failure | Eggs/red-winged blackbirds (wild population) | 810 | |
| Phytoestrogens | |||
| Genistein | Aggressive, defensive behaviors | Behavior/male/mice | 811 |
| Retention of cancellous bone after ovariectomy | Tibia bones/female/rat | 812 | |
| Expression of OPN, activation of Akt | Prostate/male/mice | 740 | |
| Resveratrol | Angiogenesis | Chorioallantoic membrane/chicken embryos | 742 |
| Ulcer index after chemical treatment, expression of gastroprotective genes | Stomach/male/mice | 813 | |
| Phytochemicals | |||
| Phlorizin | Memory retention | Behavior/male/mice | 814 |
| Herbicides | |||
| Atrazine | Time to metamorphosis | Thyroid axis/Rhinella arenarum (South American toad) | 815 |
| Survivorship patterns | Four species of frogs | 363 | |
| Growth parameters | Bufo americanus | 816 | |
| Pendimethalin | Expression of AR, IGF-I | Uterus/female/mice | 817 |
| Commercial mixture with mecoprop, 2,4-dichlorophenoxyacetic acid and dicamba | Number of implantation sites, number of live births | Female/mice | 818 |
| Simazine | Estrous cyclicity | Reproductive axis/female/rat | 819 |
| Insecticides | |||
| Permethrin | Dopamine transport | Brain/male/mice | 820 |
| Heptachlor | Dopamine transport | Brain/male/mice | 820 |
| DDT | Number of pups, sex ratios, neonatal body weight, male anogenital distance | Mice | 777 |
| Methoxychlor | Number of pups, anogenital distance (males and females), neurobehaviors (males and females) | Mice | 777 |
| Chlorpyrifos | Body weight | Male/rats | 821 |
| Antioxidant enzyme activity | Oxya chinensis (locusts) | 822 | |
| Malathion | Antioxidant enzyme activity | O. chinensis (locusts) | 822 |
| Fungicides | |||
| Carbendazim | Liver enzymes, hematology parameters | Blood and liver/male/rats | 823 |
| Chlorothalonil | Survival, immune response, corticosterone levels | Several amphibian species | 686 |
| Vinclozolin | Protein expression | Testes/male/P. scaber (isopod) | 786 |
Although NMDRCs attributable to estrogen treatment are well documented, the induction of NMDRCs is again observed to be a general feature of hormone treatment; a wide range of hormones produce these types of responses in exposed animals. Importantly, a number of pharmaceutical compounds with hormone-mimicking or endocrine-disrupting activities also produce NMDRCs. Finally, as expected from the results of cell culture experiments, chemicals with many different modes of action generate NMDRCs in treated animals.
Perhaps most striking is the range of endpoints affected, from higher-order events such as the number of viable offspring (which could be due to alterations in the reproductive tissues themselves or the reproductive axis), to behavioral effects, to altered organ weights, and to lower-order events such as gene expression. The mechanisms responsible for these nonmonotonic phenomena may be similar to those studied in cell culture systems, although additional mechanisms are likely to be operating in vivo such as alterations in tissue organization (541) and the interactions of various players in the positive and negative feedback loops of the endocrine system.
3. Examples of NMDRCs in the epidemiology literature
Perhaps not surprisingly, natural hormones produce NMDRCs in human populations as well (Table 8). Although the methods needed to detect NMDRCs in humans are specific to the field of epidemiology, these results support the idea that NMDRCs are a fundamental feature of hormones. Importantly, it should be noted that most of the individuals surveyed in studies examining the effects of natural hormones have a disease status or are elderly. This of course does not mean that natural hormones induce NMDRCs in only these select populations but may instead be a reflection of the types of individuals available for these studies (for example, there are very few clinical events in younger people).
Table 8.
NMDRCs for natural hormones identified in the epidemiology literature
| Hormone | Affected endpoint | NMDRC | Study subjects | Refs. |
|---|---|---|---|---|
| Testosterone (free) | Incidence of coronary events | Incidence of 25% at extremes of exposure, 16% at moderate exposure | Rancho Bernardo Study participants, women aged 40+ (n = 639) | 824 |
| Depression | Hypo- and hypergonadal had higher depression scores than those with intermediate free testosterone | Androx Vienna Municipality Study participants, manual workers, men aged 43–67 (n = 689) | 825 | |
| PTH | Mortality | ∼50% excess risk for individuals with low or high iPTH | Hemodialysis patients (n = 3946) | 826 |
| Risk of vertebral or hip fractures | ∼33% higher for low or high iPTH compared to normal levels | Elderly dialysis patients (n = 9007) | 827 | |
| TSH | Incidence of Alzheimer’s disease | About double the incidence in lowest and highest tertile in women (no effects observed in men) | Framingham Study participants (elderly) (n = 1864, 59% women) | 828 |
| Leptin | Mortality | Mortality ∼10% higher for lowest and highest leptin levels | Framingham Heart Study participants (elderly) (n = 818, 62% women) | 563 |
| Insulin | Coronary artery calcification | Higher for low and high insulin area under the curve measures. | Nondiabetic patients with suspected coronary heart disease, cross-sectional (n = 582) | 829 |
| Mortality (noncardiovascular only) | Relative risk ∼1.5 for highest and lowest fasting insulin levels | Helsinki Policemen Study participants, men aged 34–64 (n = 970) | 830 | |
| Cortisol | BMI, waist circumference | Low cortisol secretion per hour for individuals with highest and lowest BMI, waist circumference | Whitehall II participants, adults, cross-sectional (n = 2915 men; n = 1041 women) | 831 |
| Major depression (by diagnostic interview) | Slight increases at extremes of cortisol | Longitudinal Aging Study Amsterdam participants, aged 65+, cross-sectional (n = 1185) | 832 |
NMDRCs observed in the epidemiology literature from human populations exposed to EDCs are now starting to receive attention (Table 9). Here, most reports of NMDRCs come from studies of healthy individuals exposed to persistent organic pollutants POPs, chemicals that do not easily degrade and consequently bioaccumulate in human and animal tissues (542). These POPs do encompass a range of chemical classes including components of plastics, pesticides, and industrial pollutants. A large number of these studies have focused on endpoints that are relevant to metabolic disease, and together, these studies show that there is a recurring pattern of NMDRCs related to POPs and disease. Of course, not every study of POPs shows NMDRCs, and this is probably due to the distribution of EDCs in the populations examined.
Table 9.
NMDRCs for EDCs identified in the epidemiology literature
| Chemicals by chemical class | Affected endpoint | NMDRC | Study subjects | Refs. |
|---|---|---|---|---|
| Insecticides | ||||
| Trans-nonachlor | Diabetes incidence | Highest risk in groups with intermediate exposures (quartile 2) | CARDIA participants, case-control study (n = 90 cases and n = 90 controls) | 833 |
| Telomere length in peripheral leukocytes | Increased length in intermediate exposures (quintile 4) | Adults aged 40+ (Korea, n = 84) | 591 | |
| p,p′-DDE | BMI, triglyceride levels, HDL cholesterol | Highest risk in groups with intermediate exposures (quartile 3) | CARDIA participants (n = 90 controls from nested case control study) | 590 |
| Risk of rapid infant weight gain | For infants born to women of normal weight prepregnancy, risk is highest with intermediate exposures. | Infants from Childhood and the Environment project, Spain (n = 374 from normal prepregnancy weight mothers; n = 144 from overweight mothers) | 834 | |
| Telomere length in peripheral leukocytes | Increased length with intermediate exposures (quintile 4) | Adults aged 40+ (Korea, n = 84) | 591 | |
| Oxychlordane | Bone mineral density of arm bones | With low exposures, fat mass had inverse associations with bone mineral density; with high exposures, fat mass had positive associations with bone mineral density. | NHANES 1999–2004 participants, aged 50+ (n = 679 women, n = 612 men) | 835 |
| Plastics | ||||
| Mono-methyl phthalate (MMP) | Atherosclerotic plaques | Increased risk in intermediate exposure groups (quintiles 2–4) | Adults aged 70, living in Sweden (n = 1016) | 836 |
| Perfluorinated compounds | ||||
| PFOA | Arthritis (self-reported) | Increased risk in intermediate exposure groups (quartile 2) | NHANES participants, aged 20+ (both sexes, n = 1006) | 837 |
| Fire retardants | ||||
| PBB-153 | Blood triglyceride levels | Increased risk in intermediate exposure groups (quartile 2) | NHANES participants, aged 12+ (n = 637) | 604 |
| PBDE-153 | Prevalence of diabetes, | Prevalence of diabetes highest in intermediate groups (quartiles 2–3 relative to individuals with undetectable levels) | NHANES participants, aged 12+ (n = 1367) | 604 |
| Prevalence of metabolic syndrome, levels of blood triglycerides | Prevalence of metabolic syndrome highest in intermediate exposure groups (quartile 2 relative to individuals with undetectable levels); blood triglycerides highest in low exposure groups (quartile 1 relative to individuals with undetectable levels) | NHANES participants, aged 12+ (n = 637) | 604 | |
| PCB | ||||
| PCB-74 | Triglyceride levels | Lowest levels are observed in intermediate groups (quartile 2) | CARDIA participants (n = 90 controls from nested case-control study) | 590 |
| PCB-126 | Bone mineral density in right arm | With low exposures, fat mass had inverse associations with bone mineral density; with high exposures, fat mass had positive associations with bone mineral density | NHANES participants, aged <50 (n = 710 women, n = 768 men) | 835 |
| PCB-138 | Bone mineral density in right arm | With low exposures, fat mass had inverse associations with bone mineral density; with high exposures, fat mass had positive associations with bone mineral density | NHANES participants, women aged 50+ (n = 679 women, n = 612 men) | 835 |
| PCB-153 | Telomere length in peripheral leukocytes | Increased length with intermediate exposure groups (quintile 4) | Adults aged 40+ (Korea, n = 84) | 591 |
| PCB-170 | Diabetes incidence | Highest risk in groups with intermediate exposures (quartile 2) | CARDIA participants, case-control study (n = 90 cases and n = 90 controls) | 833 |
| Endometriosis | Decreased risk in groups with intermediate exposures (quartile 3) | Participants from the Women at Risk of Endometriosis (WREN) study, 18–49 yr old, case-control study (n = 251 cases; n = 538 controls) | 838 | |
| PCB-172 | DNA hypomethylation (by Alu assay) | Highest levels of hypomethylation in groups with lowest and highest exposures | Adults aged 40+ (Korea, n = 86) | 839 |
| PCB-180a | BMI | Highest BMI with intermediate exposures (quartile 2) | CARDIA participants (n = 90 controls from nested case control study) | 590 |
| PCB-187a | HDL cholesterol levels | Lowest levels with intermediate exposures (quartile 2) | CARDIA participants (n = 90 controls from nested case control study) | 590 |
| PCB 196–203 | Diabetes incidence | Highest risk in groups with intermediate exposures (quartile 2) | CARDIA participants, case-control study (n = 90 cases and n = 90 controls) | 833 |
| PCB-196 | Endometriosis | Decreased risk in groups with intermediate exposures (quartile 3) | Participants from the Women at Risk of Endometriosis (WREN) study, 18–49 yr old, case-control study (n = 251 cases; n = 538 controls) | 838 |
| PCB-199a | Triglyceride levels | Highest risk in groups with intermediate exposures (quartiles 2–3) | CARDIA participants (n = 90 controls from nested case control study) | 590 |
| PCB-201 | Endometriosis | Decreased risk in groups with intermediate exposures (quartiles 2–3) | Participants from the Women at Risk of Endometriosis (WREN) study, 18–49 yr old, case-control study (n = 251 cases, n = 538 controls) | 838 |
| Heavy metals | ||||
| Selenium | Fasting glucose levels (by modeled exposure) | Intermediate exposures have highest fasting glucose levels | NHANES 2003- 2004 participants, aged 40+ (n = 917) | 840 |
| Glycosylated hemoglobin (by modeled exposure) | Intermediate exposures have highest % glycosylated hemoglobin | NHANES 2003- 2004 participants, aged 40+ (n = 917) | 840 | |
| Diabetes incidence (by modeled exposure) | Intermediate exposures have highest risk for diabetes | NHANES 2003- 2004 participants, aged 40+ (n = 917) | 840 | |
| Blood triglyceride levels | Intermediate exposures have highest triglyceride levels | NHANES participants, aged 40+ (n = 1159) | 841 | |
| Arsenic | Cytokines in umbilical cord blood | Lower inflammatory markers at intermediate exposures (quartile 2) | Pregnant women in Bangladesh (n = 130) | 842 |
| Manganese | Mental development scores in infants and toddlers | Intermediate exposures had highest mental development scores at 12 months of age; association lost in older toddlers | 12-month-old infants, Mexico (n = 301) | 843 |
| Sperm count, motility and morphology | Intermediate doses had lowest sperm counts and motility; intermediate doses also had the worst sperm morphologies | Men aged 18–55 (infertility clinic patients, n = 200) | 844 | |
| Mixtures | ||||
| 31 POP | Diabetes incidence | Highest incidence in intermediate groups (sextiles 2–3) | CARDIA participants, case-control study (n = 90 cases and n = 90 controls) | 833 |
| 16 POP | Diabetes incidence | Highest incidence in intermediate groups (sextiles 2–3) | CARDIA participants, case-control study (n = 90 cases and n = 90 controls) | 833 |
| Non-dioxin-like PCB (mix) | Metabolic syndrome | Highest incidence in intermediate groups (quartile 3) | NHANES 1999–2002 participants, aged 20+ (n = 721) | 845 |
| Dioxin-like PCB (mix) | Triacylglycerol levels by quartile of exposure | Highest levels in intermediate groups (quartile 3) | NHANES 1999–2002 participants, aged 20+ (n = 721) | 845 |
| Additional supportive evidence for NMDRC in the epidemiology literature | ||||
| Insecticides | ||||
| Heptachlor epoxide | Prevalence of newly diagnosed hypertension | Highest risk in intermediate groups (quartile 2); other endpoints do not have NMDRC | NHANES participants, women aged 40+, cross-sectional (n = 51 cases, n = 278 total) | 26 |
| β-Hexachlorocyclohexane | Triacylglycerol levels by quartile of exposure | Highest risk in intermediate group (quartile 2) | NHANES participants, aged 20+ (n = 896 men, 175 with metabolic syndrome) | 845 |
| Plastics | ||||
| Mono-N-butyl phthalate (MBP) | BMI, age-specific effects | Effects seen only in elderly participants (age 60–80); risk is lowest in quartile 3 | NHANES male participants (n = 365; age 60–80) | 470 |
| Mono-benzyl phthalate (MBzP) | BMI, age-specific effects | Effects seen only in young participants (age 6–11); risk is highest in quartiles 2–3 | NHANES participants (both sexes, n = 329 males; n = 327 females) | 470 |
| Flame retardants | ||||
| PFOA | Thyroid disease (self-reported) | Lowest risk in intermediate groups (quartile 3) | NHANES 1999–2000, 2003–2006 participants, males aged 20+ (n = 3974) | 837 |
| Dioxin and related compounds | ||||
| TCDD | Age at natural menopause | Highest for intermediate exposure group (quintile 4) | Highly exposed women; Seveso Women’s Health Study participants (n = 616) | 468 |
| HCDD | Bone mineral density in right arm by quintile of fat mass | With low exposures, fat mass had inverse associations with bone mineral density; with high exposures, fat mass had positive associations with bone mineral density | NHANES participants, women aged 50+ (n = 679 women, n = 612 men) | 835 |
| Heavy metals | ||||
| Selenium | Prevalence of peripheral artery disease | Disease prevalence decreased in intermediate doses, then increased gradually with higher doses | NHANES participants, aged 40+ (n = 2062) | 469 |
In addition to the studies that show strong evidence for NMDRCs in human populations, there is also a subset of studies that provide suggestive evidence for nonmonotonic relationships between EDCs and human health endpoints (Table 9). In fact, the authors of many of these papers clearly identify U- or inverted U-shaped dose-response curves. However, when authors do not perform the appropriate statistical tests to verify the presence of a NMDRC, there is some ambiguity in their conclusions. The usual cross-sectional vs. prospective design dichotomy in epidemiology also is a factor that can influence the strength of a NMDRC, or prevent the detection of one at all. This disjunction in design is often incongruous with EDC exposure studies because we often know very little about clearance rates of the chemical, interactions with adiposity, and changes to these factors with age and gender. Yet regardless of any possible weaknesses in these studies, they provide supportive evidence that NMDRCs are observed in human populations.
Because these reports of NMDRCs in human populations are relatively new, few mechanisms have been proposed for these phenomena. Why would risk curves be nonmonotonic over the dose distribution observed in human populations? Why would individuals with the highest exposures have less severe health outcomes compared with individuals with more moderate exposures? One plausible explanation is that the same mechanisms for NMDRCs in animals and cell cultures operate in human populations: chronic exposures to high doses can activate negative feedback loops, activate receptors that promote changes in different pathways that diverge on the same endpoint with opposing effects, or produce some measure of toxicity. Accidental exposures of very large doses may not behave the same as background doses for a variety of reasons, including the toxicity of high doses; these large doses tend to occur over a short time (and therefore more faithfully replicate what is observed in animal studies after controlled administration).
Another explanation is that epidemiology studies, unlike controlled animal studies, examine truly complex mixtures of EDCs and other environmental chemicals. Some chemical exposures are likely to be correlated due to their sources and their dynamics in air, water, soil, and living organisms that are subsequently eaten. Therefore, intake of these chemicals may produce unpredicted, likely nonlinear outcomes whether the two chemicals act via similar or different pathways.
The design of observational epidemiological studies is fundamentally different from studies of cells or animals, in that the EDC exposure distributions are given, rather than set by the investigator. In particular, as shown in Fig. 5, different epidemiological populations will have different ranges of exposure, with the schematic example showing increasing risk in a population with the lowest exposures (labeled group A), an inverted U-shaped risk in a moderate dose population (labeled group B), and an inverse risk in a population with the highest exposures (labeled group C). An additional example is provided (labeled group D) in which an industrial spill shows high risk, but the comparison with the entire unaffected population with a wide variety of risk levels due to differential background exposure could lead to a high- or a low-risk reference group and a wide variety of possible findings.

It is reasonable to suggest that even though epidemiological studies are an assessment of exposures at a single time point, many of these pollutants are persistent, and therefore a single measure of their concentration in blood may be a suitable surrogate for long-term exposures. The movement of people from relatively low- to higher-exposure groups over time depend on refreshed exposures, clearance rates, and individual differences in ability to handle exposures (i.e. due to genetic susceptibilities, amount of adipose tissue where POPs can be stored, etc.).
Figure 5 therefore further illustrates that observational epidemiological studies yield the composite effect of varying mixtures of EDCs at various exposure levels for various durations, combining acute and chronic effects. These studies are important, however, in that they are the only way to study EDC effects in the long term in intact humans, as opposed to studying signaling pathways, cells, organs, or animal models over limited periods of time. Causal inference is not done directly from the epidemiological study results; instead, it is done via combining information from the epidemiological observations with findings from the detailed studies of pathways and animals.
We have suggested that NMDRCs are a fundamental and general feature of hormone action in cells and animals. It is therefore worth asking whether NMDRCs are expected in the epidemiology literature. The endpoints assessed in epidemiology studies are typically integrated effects, rather than short-term effects; therefore, the various cell- or organ-specific effects may cancel each other, particularly if they are NMDRCs (because they are unlikely to all have nonmonotonicity at the same dose and direction). Thus, NMDRCs are likely to be rarer in the epidemiology literature compared with studies examining the effects of a wide range of doses of an EDC on animals and cultured cells. Yet it is also important to ask what can be concluded if a NMDRC is detected in one epidemiology study but not in others examining the same chemical and outcome. There are several factors that must be considered. The first is that differences in the populations examined between the two studies could explain why a monotonic relationship is observed in one group and a nonmonotonic relationship in another (see Fig. 5). The second is that one or more studies may not be statistically designed to detect NMDRCs. Finally, it is plausible that the NMDRC is an artifact due to residual confounding or some other factor that was not considered in the experimental design. As more becomes known about the mechanisms operating in cells, tissues, and organs to generate NMDRCs, our ability to apply this information to epidemiology studies will increase as well.
4. Tamoxifen flare, a NMDRC observed in cells, animals, and human patients
Although there is controversy in toxicology and risk assessment for endocrine disruptors, NMDRCs are recognized and used in current human clinical practice, although under a different specific term, flare. Flare is often reported in the therapy of hormone-dependent cancers such as breast and prostate cancer. Clinically, failure to recognize the NMDRC that is termed a flare would be considered malpractice in human medicine.
Tamoxifen flare was described and named as a transient worsening of the symptoms of advanced breast cancer, particularly metastases to bone associated with increased pain, seen shortly after the initiation of therapy in some patients (543). If the therapy could be continued, the patients showing tamoxifen flare demonstrated a very high likelihood of subsequent response to tamoxifen, including arrest of tumor growth and progression of symptoms for some time.
The subsequent mechanism of the flare was described in basic lab studies in athymic mouse models of human hormone-dependent breast cancer xenografts (544) and in tissue culture of hormone-dependent human breast cancer cells (545–547). In these models, it was observed that although high, therapeutic concentrations of tamoxifen inhibited estrogen-stimulated proliferation of breast cancer cells, lower concentrations of tamoxifen actually stimulated breast cancer cell growth as long as the cells were estrogen dependent (548). Tamoxifen was also shown to disrupt tissue organization of the mammary gland, with specific effects on the stroma that may contribute to the observed effects on proliferation of epithelial cells (549, 550).
Tamoxifen therapy is administered as 10 mg twice per day (20 mg/d; approx 0.3 mg/kg body weight per day), but the target circulating levels are in the near submicromolar range (0.2–0.6 μm); these levels are reached slowly, after approximately 2 wks of therapy (551). In the initial period, where tamoxifen flare is observed, the circulating concentrations are ascending through lower concentrations, in the range below therapeutic suppression of growth, where breast cancer cell proliferation is actually stimulated by the drug, both in tissue culture, in animal xenograft studies, and in human patients (reviewed in Ref. 548). The recognition of this dual dose-response range for tamoxifen (low-dose, low-concentration estrogenic growth-stimulatory and higher-dose, higher-concentration estrogenic growth-inhibitory responses) led to the definition of the term selective estrogen response modulator, or SERM, activity (552–554). This SERM activity has since been observed for many or even most estrogenic EDCs, including BPA (3, 555–557).
These observations defined three separate dose-response ranges for the SERM tamoxifen in human clinical use. The lowest dose-response range, the range of flare, stimulated breast cancer growth and symptoms in some patients with hormone-dependent cancer. The next higher dose-response range is the therapeutic range where tamoxifen inhibits estrogen-dependent tumor growth. The highest dose range causes acute toxicity by the SERM (see Fig. 6).

Tamoxifen provides an excellent example for how high-dose testing cannot be used to predict the effects of low doses. For tamoxifen (as for other drugs), the range of acute human toxicity for tamoxifen was determined in phase I clinical trials. Phase I trials also defined an initial therapeutic range, the second dose-response range, as a dose below which acute toxicity was not observed. The therapeutic dose range was tested and further defined in phase II and later clinical trials to determine efficacy (see for example Ref. 558). Standard toxicological testing from high doses to define a LOAEL or NOAEL are equivalent to the phase I clinical testing, and in risk assessment, a safe dose or reference dose is calculated from these tests. However, the lowest dose range, with the highly adverse effects termed flare, was not detected in the phase I trials and was determined only for tamoxifen in breast cancer therapy at the therapeutic doses (543). The implication for risk assessment is that NMDRCs for EDCs, particularly those already identified as SERMs, would likely not be detected by standard toxicological testing at high doses. That is, the consequence of high-dose testing is the calculation of a defined but otherwise untested safe dose that is well within the range equivalent to flare, i.e. a manifestly unsafe dose of the EDC (Fig. 6).
5. Similarities in endpoints across cell culture, animal, and epidemiology studies: evidence for common mechanisms?
There are common trends in some findings of NMDRCs in cell, animal, and human studies and therefore evidence for related mechanisms for NMDRCs at various levels of biological complexity. Tamoxifen flare, discussed in Section III.C.4, is an informative example. Another illustrative example is that of the effect of the hormone leptin (Fig. 7). In cultured primary adipocytes, NMDRCs are observed after leptin exposure; moderate doses of leptin significantly reduce insulin-mediated glucose intake, whereas low and high doses maintain higher glucose intake in response to insulin (559). The rat pancreas shows a similar response to leptin; the amount of secreted insulin has an inverted U-shaped response to leptin (560, 561). Even more striking is the relationship between leptin and food intake. Rats administered moderate doses of leptin consume less food compared to rats dosed with low or high levels of leptin (562); mechanistically, this lower food intake could be due to higher circulating glucose levels in these animals due to ineffective insulin action. And finally, in a human study, leptin levels were found to correlate with body mass index but have a U-shaped relationship with mortality (563). These results suggest that hormones can produce similar responses at several levels of biological complexity (cell, organ, animal, and population).

A large number of epidemiology studies with NMDRCs have found relationships between EDC exposures like POPs and metabolic diseases including obesity and diabetes (Table 9) (see also Ref. 564 for a review), and the mechanisms for these relationships have begun to be explored. Human and animal cells treated with EDCs in culture display NMDRCs that are relevant to these diseases: BPA has nonmonotonic effects on the expression of adipocyte proteins in preadipocytes and the release of adiponectin from mature adipocytes (565–567). Similarly, in female rodents, low doses but not high doses of BPA increased adipose tissue weight and serum leptin concentrations (568), and intermediate doses of phthalates decrease serum cholesterol levels (569). Thus, although understanding the mechanisms operating at the cellular level of organization has not yet led to definitive knowledge of the mechanisms producing NMDRCs in human populations, there appear to be strong similarities in cells, animals, and humans that support a call for continued work focusing on metabolic disease endpoints at each level of biological organization.
D. NMDRC summary
We have demonstrated that nonmonotonicity is a common occurrence after exposures to hormones and EDCs in cell culture and animals and across human populations. Because of the abundance of examples of NMDRCs, we expect that if adequate dose ranges are included in animal and cell culture studies, including the use of negative and well-chosen positive controls, NMDRCs may be observed more often than not. Here, we have focused mainly on studies that examined a wide range of doses, including many that examined the effects of doses that span the low-dose and toxicological ranges. We also discussed several mechanisms that produce NMDRCs. Each of these mechanisms can and does operate at the same time in a biological system, and this cooperative action is ultimately responsible for NMDRCs.
Understanding nonmonotonicity has both theoretical and practical relevance. When a chemical produces monotonic responses, all doses are expected to produce similar effects whose magnitude varies with the dose, but when a chemical produces a NMDRC, dissimilar or even opposite effects will be observed at different doses. Thus, monotonic responses can be modeled using the assumption that each step in a linear pathway behaves according to the law of mass action (43, 570); high doses are always expected to produce higher responses. In contrast, NMDRCs are not easy to model (although they are quite easy to test for), requiring detailed knowledge of the specific mechanisms operating in several biological components. From a regulatory standpoint, information from high doses cannot always be used to assess whether low doses will produce a biological effect (38).
IV. Implications of Low-Dose Effects and Nonmonotonicity
Both low-dose effects and NMDRCs have been observed for a wide variety of EDCs as well as natural hormones. Importantly, these phenomena encompass every level of biological organization, from gene expression, hormone production, and cell number to changes in tissue architecture to behavior and population-based disease risks. One conclusion from this review is that low-dose effects and NMDRCs are often observed after administration of environmentally relevant doses of EDCs. For both hormones and EDCs, NMDRCs should be the default assumption absent sufficient data to indicate otherwise. Furthermore, there are well-understood mechanisms to explain how low-dose effects and NMDRCs manifest in vitro and in vivo. Accepting these phenomena, therefore, should lead to paradigm shifts in toxicological studies and will likely also have lasting effects on regulatory science. Some of these aspects are discussed below. Additionally, we have briefly explored how this knowledge should influence future approaches in human and environmental health.
At a very practical level, we recommend that researchers publishing data with low-dose and nonmonotonic effects include key words in the abstract/article that identify them as such specifically. This review was unquestionably impeded because this has not been standard practice. We also strongly recommend that data showing nonmonotonic and binary response patterns not be rejected or criticized because there is no dose response.
A. Experimental design
1. Dose ranges must be chosen carefully
To detect low-dose effects or NMDRCs, the doses included for testing are of utmost importance. Most of the studies we examined here for nonmonotonicity tested doses over severalfold concentrations. Unfortunately, regulatory guidelines only require that three doses be tested. Both low-dose effects and NMDRCs can be observed when examining only a few doses, but some studies may detect significant results purely by luck, because a small shift in dose can have a large impact on the ability to observe differences relative to untreated controls.
In the multitude of chemicals that have never been tested at low doses, or in the development of new chemicals, to determine whether a chemical has low-dose effects in laboratory animals, we suggest setting the NOAEL or LOAEL from traditional toxicological studies as the highest dose in experiments specifically designed to test endocrine-sensitive endpoints. We suggest setting the lowest dose in the experiment below the range of human exposures, if such a dose is known. Several intermediate doses overlapping the range of typical human exposures should be included also, bringing the total number in the range of five to eight total doses tested. Importantly, although the levels of many environmental chemicals in human blood and/or urine have been reported by the CDC and other groups responsible for population-scale biomonitoring, it is often not known what administered doses are needed to achieve these internal exposure levels in animals (4, 253); thus, toxicokinetic studies are often needed before the onset of low-dose testing. This is important because the critical issue is to determine what effects are observed in animals when circulating levels of an EDC match what is measured in the typical human. Due to differences in metabolism, route of exposure, and other factors, a relatively high dose may need to be administered to a rodent to produce blood concentrations in the range of human levels; however, this should not be considered a high-dose study.
It has also been suggested that animal studies that are used to understand the potential effects of a chemical on humans should use a relevant route of administration to recapitulate human exposures (571, 572) because there may be differences in metabolism after oral and nonoral administration. Many chemicals that enter the body orally undergo first-pass metabolism and are then inactivated via liver enzymes, whereas other routes (i.e. sc) can bypass these mechanisms and lead to a higher concentration of the active compound in circulation (573). Studies indicate, however, that inactivation of chemicals via first-pass metabolism is not complete and also that deconjugation of metabolites can occur in some tissues allowing the re-release of the active form (574, 575). Additionally, for some chemicals, it is clear that route of administration has little or no impact on the availability of the active compound in the body (241, 384), and other studies show that route of administration has no impact on the biological effects of these chemicals; i.e. regardless of how it enters the body, dioxin has similar effects on exposed individuals (384), and comparable results have been observed for BPA (141). Although understanding the typical route of human exposure to each environmental chemical is an important task, it has been argued that any method that leads to blood concentrations of a test chemical in the range they are observed in humans is an acceptable exposure protocol, and this is especially true with gestational exposures, because fetuses are exposed to chemicals only via their mothers’ blood (31, 576).
2. Timing of exposures is important
Rodent studies indicate that EDC exposures during development have organizational effects, with permanent effects that can manifest even in late adulthood, whereas exposures after puberty are for the most part activational, with effects that are abrogated when exposures cease. For example, the adult uterus requires relatively large doses of BPA (in the parts-per-million range) to induce changes associated with the uterotrophic assay (555, 577), whereas parts-per-trillion and ppb exposures during the fetal period permanently and effectively alter development of the uterus (279, 310, 578). Thus, the timing of exposures is profoundly important to detect low-dose effects of EDCs.
Human studies also support this conclusion. The 1976 explosion of a chemical plant in Seveso, Italy, which led to widespread human exposure to large amounts of TCDD, a particularly toxic form of dioxin, and the deposition of this chemical on the land surrounding the chemical plant, provided evidence in support of the organizational and activational effects of endocrine-active chemicals in humans (579). Serum TCDD concentrations showed correlations between exposure levels and several disease outcomes including breast cancer risk, abnormal menstrual cycles, and endometriosis (580–582), but individuals who were either infants or teenagers at the time of the explosion were found to be at greatest risk for developing adult diseases (583, 584). Importantly, many scientists have argued that organizational effects can occur during puberty, i.e. that the period where hormones have irreversible effects on organ development extends beyond the fetal and neonatal period (585), and for some endpoints this appears to be the case (586, 587).
It has also been proposed that the endocrine system maintains homeostasis in the face of environmental insults (210). The adult endocrine system does appear to provide some ability to maintain a type of homeostasis; when the pharmaceutical estrogen DES is administered to pregnant mice, the circulating estradiol concentrations in the dam respond by decreasing linearly (224). In contrast, fetal concentrations of estradiol respond nonmonotonically in a way that is clearly not correlated with maternal levels. Similarly, there is evidence that BPA can induce aromatase and therefore increase estradiol levels in situ in the fetal urogenital sinus (588). This is an example of a feed-forward positive-feedback effect rather than a homeostatic response. The effects of EDCs on adult subjects, both animal and people, suggest that diseases often result from low-dose adult exposures (589–595); this argues against a view of the endocrine system as a means to maintain homeostatic control. Instead, individuals can be permanently changed, in an adverse way, after EDC exposures.
In one example, pregnant mice were exposed to low concentrations of BPA, and their male offspring had altered pancreatic function at 6 months of age (158). Surprisingly, however, the mothers (exposed only during pregnancy) were also affected, with altered metabolic machinery and body weight at 4 months postpartum, long after exposures had ended. The increased incidence of breast cancer in women that took DES during pregnancy also illustrates this point (596, 597). These studies suggest that even the adult endocrine system is not invariably capable of maintaining a so-called homeostatic state when exogenous chemicals affecting the endocrine system are present. Thus, although adult exposures to EDCs have been given some attention by bench scientists (29), more work of this kind is needed to better understand whether and how EDCs can have permanent organizational effects on adult animals.
At the beginning of this review, we justified the need to critically examine the low-dose literature because of recent epidemiological findings linking EDC exposures and diseases. Yet there is inherent difficulty in examining neonatal exposures to EDCs and their connection to diseases due to the length of time needed for these studies; thus, many studies of this type have examined high doses of pharmaceuticals (i.e. DES) or accidental exposures to industrial chemicals (i.e. dioxin) (66, 398, 399, 581, 597–601).
Only recently, with the availability of biomonitoring samples from large reference populations, have lower doses begun to receive widespread attention from epidemiologists. Many recent studies have examined adult exposures to EDCs and correlated exposures with disease statuses (see for example Refs. 15, 16, and 602–604). Human studies examining fetal/neonatal exposures to low-dose EDCs and early life effects have also begun to be studied (6, 333, 605–607), although studies linking these early life exposures to adult diseases are likely to be decades away. More than anything, these studies support our view that the effects of low-dose exposures should be considered when determining chemical safety.
3. Importance of endpoints being examined
Traditional toxicology testing, and in particular those studies performed for the purposes of risk assessment, typically adhere to guideline studies that have been approved by international committees of experts (608). The endpoints assessed in these guideline-compliant studies are centered around higher-order levels, including weight loss, mortality, changes in organ weight, and a limited number of histopathological analyses (609, 610). When pregnant animals are included in toxicological assessments, the endpoints measured typically include the ability to maintain pregnancies, the number of offspring delivered, sex ratios of surviving pups, and measures regarding maternal weight gain and food/water intake (610).
Yet low-dose EDCs are rarely toxic to the point of killing adult animals or causing spontaneous abortions, and traditional tests such as the uterotrophic assay have been shown to be relatively insensitive (72, 577). It has been argued that this type of testing is insufficient for understanding the effects of EDCs (31, 70, 495, 611). Many EDC studies have instead focused on examining newly developed, highly sensitive endpoints that span multiple levels of biological organization, from gene expression to tissue organization to organ systems to the whole animal (612), which may not be rapidly lethal but which nonetheless have enormous importance for health, including mortality. Thus, for example, studies designed to examine the effects of chemicals on obesity no longer focus on body weight alone but also analyze gene expression; fat content in adipose cells and the process of adipogenesis; inflammation, innvervation, and vascularization parameters in specific fat pads; conversion rates of white and brown adipose tissues; systemic hormone levels and response to glucose and insulin challenges; and food intake and energy expenditures, among others (314, 613–615). As our knowledge of EDCs and the endocrine system continue to grow, the most sensitive endpoints should be used to determine whether a chemical is disrupting the development of organisms (70).
In moving beyond traditional, well-characterized health-related endpoints like mortality and weight loss, an important question has been raised: how do we define endpoints as adverse? This is an important point, because it has been suggested that the endpoints examined in independent EDC studies are not validated and may not represent adverse effects (609). There is also debate over whether the mechanism (or mode) of action must be explained for each effect to determine whether a relevant pathway is present in humans (616, 617). Yet, when originally assessing the low-dose literature, the NTP expert panel chose to examine all effects of EDC exposure, regardless of whether the endpoint could be deemed adverse (2). From the perspective of developmental biology, any change in development should be seen as adverse, even if the change itself is not associated with a disease or dysfunction. Some of these developmental changes, in fact, may increase sensitivity or susceptibility to disease later on in life but will otherwise appear normal. Furthermore, studies of heavy metals have shown that small shifts in parameters like IQ may not have drastic effects on individuals but can have serious repercussions on the population level (618), and therefore changes in the variance/observable range of a phenotype should also be considered adverse (52).
4. Importance of study size
National Institutes of Health guidelines require that the number of vertebrate animals used in experiments be as small as possible to show statistically significant effects based on power analysis. Yet many traditional toxicology studies have used large numbers of animals to draw conclusions about chemical safety. When the endpoints being assessed have binary outcomes (i.e. animal has a tumor vs. animal does not have a tumor) and the incidence of the phenotype is not high, a large number of animals is required to reveal statistically significant effects. In contrast, many of the endpoints examined in the field of endocrine disruption are more complex and are not binary; thus, power analysis allows researchers to determine how many animals are needed to observe statistically significant (and biologically relevant) differences between control and exposed populations. For this reason, arbitrary numbers set as cutoffs for determining whether a study is acceptable or unacceptable for risk assessments are not appropriate. Instead, the number of animals required for a study to be complete is dependent on the effect size, precision/variance, minimal meaningful difference to be considered between populations, and the α-value set in statistical tests.
B. Regulatory science
For decades, regulatory agencies have tested, or approved testing, of chemicals by examining high doses and then extrapolating down from the NOAEL, NOEL, and LOAEL to determine safe levels for humans and/or wildlife. As discussed earlier, these extrapolations use safety factors that acknowledge differences between humans and animals, exposures of vulnerable populations, interspecies variability, and other uncertainty factors. These safety factors are informed guesses, not quantitatively based calculations. Using this traditional way of setting safe doses, the levels declared safe are never in fact tested. Doses in the range of human exposures are therefore also unlikely to be tested. This has generated the current state of science, where many chemicals of concern have never been examined at environmentally relevant low doses (see Table 4 for a small number of examples).
Assumptions used in chemical risk assessments to estimate a threshold dose below which daily exposure to a chemical is estimated to be safe are false for EDCs. First, experimental data provide evidence for the lack of a threshold for EDCs (619). More broadly, the data in this review demonstrate that the central assumption underlying the use of high doses to predict low-dose effects will lead to false estimates of safety. The use of only a few high doses is based on the assumption that all dose-response relationships are monotonic and therefore that it is appropriate to apply a log-linear extrapolation from high-dose testing to estimate a safe reference dose (Fig. 4). The Endocrine Society issued a position statement on EDCs (620) and urged the risk assessment community to use the expertise of their members to develop new approaches to chemical risk assessments for EDCs based on principles of endocrinology. Undertaking this mission will represent a true paradigm shift in regulatory toxicology (79). The Endocrine Society statement was then supported in March 2011 by a letter to Science from eight societies with relevant expertise representing over 40,000 scientists and medical professionals (621).
Studies conducted for the purposes of risk assessment are expected to include three doses: a dose that has no effects on traditional toxicological endpoints (the NOAEL), a higher dose with effects on traditional endpoints (the LOAEL), and an even higher dose that shows toxicity. Although reducing the number of animals used for these types of studies is an important goal, more than three doses are often needed for a true picture of a chemical’s toxicity. The examination of a larger number of doses would allow for 1) the study of chemicals at the reference dose, i.e. the dose that is calculated to be safe; 2) examination of doses in the range of actual human exposures, which is likely to be below the reference dose; and 3) the ability to detect NMDRCs, particularly in the low-dose range. The impact of testing more doses on the numbers of animals required can be mitigated by use of power analysis, as suggested above. Because no amount of research will ever match the diversity and reality of actual human experience, there should be ongoing epidemiological study of potential adverse effects of EDCs even after safe levels are published, with periodic reevaluation of those safe levels.
One issue that has been raised by regulatory agencies is whether animal models are appropriate for understanding the effects of EDCs on humans. These arguments largely center around observed differences in hormone levels during different physiological periods in rodents and humans (57), and differences in the metabolic machinery and excretion of chemicals between species (622). To address the first issue, it should be noted that the FDA uses animals to test pharmaceuticals and other chemicals before any safety testing in humans because it is widely recognized that, although animals and humans do not have exactly the same physiologies, there is evolutionary conservation among vertebrates and specifically among mammals (62). Furthermore, animal studies proved to be highly predictive of the effects of DES on women, indicating that rodents are sufficiently similar to humans to reliably forecast affected endpoints in the endocrine system (64, 623). Thus, the default position must be that animal data are indicative of human effects until proven otherwise.
With regard to the second issue, BPA researchers in particular have examined species-specific differences in metabolism of this EDC. Interestingly, the pharmacokinetics of BPA in rodents, monkeys, and humans appear to be very similar (624), and regulatory agencies have subsequently concluded that rodents are appropriate models to assess the effects of this chemical (625, 626). Thus, researchers should select animal models that are sensitive to low doses of hormones and select appropriate species for the endpoints of interest. As the scope of our knowledge has broadened about how chemicals can alter the endocrine system, well beyond estrogens, androgens, and the thyroid, it is imperative that considerable thought be given to how to apply this for regulatory purposes.
C. Human health
As discussed several times throughout this review, there is now substantial evidence that low doses of EDCs have adverse effects on human health. Thus, although many epidemiological studies originally focused on occupationally exposed individuals and individuals affected by accidental exposures to high doses of environmental chemicals, these recent studies have suggested wide-ranging effects of EDCs on the general population.
Importantly, human exposures are examples of true mixtures; dozens if not hundreds of environmental chemicals are regularly detected in human tissues and fluids (91), yet very little is known about how these chemicals act in combination (627). Several studies indicate that EDCs can have additive or even synergistic effects (143, 323, 628–630), and thus these mixtures are likely to have unexpected and unpredictable effects on animals and humans. The study of mixtures is a growing and complex field that will require considerable attention in the years ahead as knowledge of EDCs in the laboratory setting are applied to human populations (631, 632).
How much will human health improve by testing chemicals at low, environmentally relevant doses and using the results to guide safety determinations? Current testing paradigms are missing important, sensitive endpoints; because they are often unable to detect NMDRCs, they cannot make appropriate predictions about what effects are occurring at low doses. At this time, it is not possible to quantify the total costs of low-dose exposures to EDCs. However, current epidemiology studies linking low-dose EDC exposures to a myriad of health problems, diseases, and disorders suggest that the costs of current low-dose exposures are likely to be substantial.
The weight of the available evidence suggests that EDCs affect a wide range of human health endpoints that manifest at different stages of life, from neonatal and infant periods to the aging adult. As the American population ages, healthcare costs continue to rise, and there are societal costs as well, with decreased quality of life concerns, decreases in work productivity due to illness or the need for workers to care for affected family members, and the psychological stresses of dealing with some outcomes like infertility. Thus, it is logical to conclude that low-dose testing, followed by regulatory action to minimize or eliminate human exposures to EDCs, could significantly benefit human health. This proposal effectively calls for greatly expanded research to give human communities feedback about themselves. It emanates from a view that human society benefits greatly from the many chemical compounds it uses but that extensive epidemiological surveillance and other focused research designs are needed to assure that the balance of risk/benefit from those chemicals is acceptable.
How much would human health benefit by a reduction in the use of EDCs? For some chemicals, minor changes in consumer habits or industrial practices can have drastic effects on exposures (633–636). Other chemicals like DDT that have been regulated in the United States for decades continue to be detected in human and environmental samples; the persistent nature of many of these agents suggests they may impact human health for decades to come. Even less-persistent chemicals like BPA are likely to remain in our environment long after a ban is enacted because of the large amounts of plastic waste leaching BPA (and other estrogenic compounds) from landfills into water sources (637) and its presence on thermal receipt paper and from there into recycled paper (638–640). Yet, despite these challenges, reducing human exposure to EDCs should be a priority, and one way to address that priority is to decrease the production and use of these chemicals. The Endocrine Society has called for such a reduction and the use of the precautionary principle, i.e. action in the presence of concerning information but in the absence of certainty to eliminate or cut the use of questionable chemicals even when cause-effect relationships are not yet established (620).
D. Wildlife
Much of the recent focus on EDCs has been on the impact of these chemicals on human health. Yet the earliest studies of EDCs that focused on the impact of these chemicals on wildlife should not be forgotten. Rachel Carson’s work on DDT and other pesticides provided some of the earliest warning signs that there were unintended consequences of chemical use. Carson’s work was ahead of its time; she understood that exceedingly small doses of these chemicals produced adverse effects, that the timing of exposures was critical, and that chemical mixtures produced compounded effects (641). Now, decades after some of the most dangerous EDCs have been regulated, they continue to be measured in environmental samples as well as the bodies of wildlife animals.
Furthermore, it should be pointed out that humans, like wildlife, are not insulated from the environment, and effects in wildlife, including nonmammalian species, are indicative of and mirror effects in humans. For example, BPA has estrogen-like effects in fish (642–644), amphibians (645, 646), and reptiles (647, 648). A recent review showed that demasculinizing and feminizing effects of atrazine have been demonstrated in fish, amphibians, reptiles, birds, and mammals, i.e. every vertebrate class examined (326); and in fact, the first report to suggest that atrazine induced aromatase was conducted in reptiles (649). Similarly, perchlorate affects fish (650–653), amphibians (654–658), and birds (659–661) via mechanisms consistent with those described for humans, and some of the earliest reports on perchlorate’s effects on thyroid function were conducted in amphibians (661, 662). Finally, ecological studies of dioxin and dioxin-like chemicals reveal effects on a range of exposed wildlife including birds (663, 664), fish (665, 666), and invertebrates (667). Although these studies have highlighted some of the species-specific effects of dioxin (389), and orders of magnitude differences in toxic equivalency factors between species (668), they also indicate the conservation of mechanisms for the effects of dioxin on a range of biological endpoints in wildlife, laboratory animals, and humans (384). In fact, in many cases, nonmammalian species are much more sensitive to EDC effects, and wildlife species serve as sentinels for environmental and public health (669–673). Thus, the effects of these chemicals on wildlife populations are likely to continue; for this reason, the low-dose effects of these chemicals are particularly worth understanding (674, 675).
V. Summary
In conclusion, we have provided hundreds of examples that clearly show that NMDRCs and low-dose effects are common in studies of hormones and EDCs. We have examined each of these issues separately and provided mechanistic explanations and examples of both. These topics are related, but they must be examined individually to be understood. The concept of nonmonotonicity is an essential one for the field of environmental health science because when NMDRCs occur, the effects of low doses cannot be predicted by the effects observed at high doses. In addition, the finding that chemicals have adverse effects on animals and humans in the range of environmental exposures clearly indicates that low doses cannot be ignored.
In closing, we encourage scientists and journal editors to publish data demonstrating NMDRCs and low-dose effects, even if the exact mechanism of action has not yet been elucidated. This is important because the study of EDC is a growing specialty that crosses many scientific fields, and scientists that work on or regulate EDCs should appreciate and acknowledge the existence of NMDRCs and low-dose effects and have access to this important information. We further recommend greatly expanded and generalized safety testing and surveillance to detect potential adverse effects of this broad class of chemicals. Before new chemicals are developed, a wider range of doses, extending into the low-dose range, should be fully tested. And finally, we envision that the concepts and empirical results we have presented in this paper will lead to many more collaborations among research scientists in academic and government laboratories across the globe, that more and more sophisticated study designs will emerge, that what we have produced herein will facilitate those making regulatory decisions, that actions taken in light of this information will begin to abate the use of EDCs, and ultimately that health impacts in people and in wildlife will be averted.
Acknowledgments
We thank many colleagues in the fields of endocrine disruption and environmental health science for fruitful discussions on the topics covered in this manuscript. We also thank three anonymous reviewers whose comments and suggestions greatly improved this review.
This work was supported by National Institutes of Health (NIH) Grants GM 087107; (to L.N.V.), ES 08314; (to A.M.S.), ES 010026; (to R.T.Z.), ES018764; (to F.S.v.S.), HL 53560; (to D.R.J.), UMC MO-VMFC0018; (to W.V.W.), a Susan G. Komen for Cure Grant FAS0703860 (to T.S.), grants from the Mitchell Kapor Foundation, the Cornell-Douglas Foundation, and the Wallace Global Fund (to T.B.H.) and a grant from the Kendeda Foundation (to J.P.M.). This article may be the work product of an employee or group of employees of the National Institute of Environmental Health Sciences (NIEHS), NIH; however, the statements, opinions or conclusions contained therein do not necessarily represent the statements, opinions or conclusions of NIEHS, NIH, or the U.S. government.
We dedicate this manuscript to Professor Howard A. Bern. Dr. Bern was an exceptionally brilliant biologist and a generous and inspiring colleague. His work spanning a wide range of organisms addressed multiple aspects of organismal and evolutionary biology. He was one of the founders of the field of comparative endocrinology and a pioneer in the study of endocrine disruption, anticipating the deleterious effects of developmental exposure to estrogens one decade before the discovery of the effects of diethylstilbestrol in women fetally exposed to this chemical. His pioneering work included, among other subjects, neuroendocrinology, reproduction, and mammary cancer. He was also an excellent mentor to many researchers who, in turn, advanced these endeavors. He left an indelible mark on all of us that had the privilege of meeting him.
Disclosure Summary: Fred vom Saal worked as a consultant and provided expert testimony (<$10K). The authors have nothing to disclose.
Footnotes
Abbreviations:
A4 Androstenedione
Ah Raryl hydrocarbon receptor
BPA bisphenol A
CDC Centers for Disease Control and Prevention
DDE dichlorodiphenyldichloroethylene
DDT dichlorodiphenyltrichloroethane
DES diethylstilbestrol
EDC endocrine-disrupting chemical
EPA Environmental Protection Agency
E Restrogen receptor
FDA Food and Drug Administration
GLP good laboratory practices
LOAEL lowest observed adverse effect level
mER membrane-associated ER
NHANES National Health and Nutrition Examination Survey
NIS sodium/iodide symporter
NMDRC nonmonotonic dose-response curve
NOEL no observed effect level
NOAEL no observed adverse effect level
NTP National Toxicology Program
PIN prostatic intraepithelial neoplasias
POP persistent organic pollutants
ppb parts per billion
SERM selective ER modulator
TCDD 2,3,7,8-tetrachlorodibenzo-p-dioxin
WoE weight of evidence
References
1. National Toxicology Program 2001. National Toxicology Program’s report of the endocrine disruptors low dose peer review. Research Triangle Park, NC: National Institute of Environmental Health Sciences [Google Scholar]
2. Melnick R, Lucier G, Wolfe M, Hall R, Stancel G, Prins G, Gallo M, Reuhl K, Ho SM, Brown T, Moore J, Leakey J, Haseman J, Kohn M. 2002. Summary of the National Toxicology Program’s report of the endocrine disruptors low-dose peer review. Environ Health Perspect 110:427–431 [PMC free article] [PubMed] [Google Scholar]
3. Welshons WV, Nagel SC, vom Saal FS. 2006. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology 147:S56–S69 [PubMed] [Google Scholar]
4. Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. 2007. Human exposure to bisphenol A (BPA). Reprod Toxicol 24:139–177 [PubMed] [Google Scholar]
5. Brucker-Davis F, Thayer K, Colborn T. 2001. Significant effects of mild endogenous hormonal changes in humans: considerations for low-dose testing. Environ Health Perspect 109:21–26 [PMC free article] [PubMed] [Google Scholar]
6. Braun JM, Yolton K, Dietrich KN, Hornung R, Ye X, Calafat AM, Lanphear BP. 2009. Prenatal bisphenol A exposure and early childhood behavior. Environ Health Perspect 117:1945–1952 [PMC free article] [PubMed] [Google Scholar]
7. Meeker JD, Barr DB, Hauser R. 2009. Pyrethroid insecticide metabolites are associated with serum hormone levels in adult men. Reprod Toxicol 27:155–160 [PMC free article] [PubMed] [Google Scholar]
8. Weuve J, Hauser R, Calafat AM, Missmer SA, Wise LA. 2010. Association of exposure to phthalates with endometriosis and uterine leiomyomata: findings from NHANES, 1999–2004. Environ Health Perspect 118:825–832 [PMC free article] [PubMed] [Google Scholar]
9. Meeker JD, Sathyanarayana S, Swan SH. 2009. Phthalates and other additives in plastics: human exposure and associated health outcomes. Philos Trans R Soc Lond B Biol Sci 364:2097–2113 [PMC free article] [PubMed] [Google Scholar]
10. Swan SH. 2008. Environmental phthalate exposure in relation to reproductive outcomes and other health endpoints in humans. Environ Res 108:177–184 [PMC free article] [PubMed] [Google Scholar]
11. Akinbami LJ, Lynch CD, Parker JD, Woodruff TJ. 2010. The association between childhood asthma prevalence and monitored air pollutants in metropolitan areas, United States, 2001–2004. Environ Res 110:294–301 [PubMed] [Google Scholar]
12. Stillerman KP, Mattison DR, Giudice LC, Woodruff TJ. 2008. Environmental exposures and adverse pregnancy outcomes: a review of the science. Reprod Sci 15:631–650 [PubMed] [Google Scholar]
13. Grün F. 2010. Obesogens. Curr Opin Endocrinol Diabetes Obes 17:453–459 [PubMed] [Google Scholar]
14. Soto AM, Sonnenschein C. 2010. Environmental causes of cancer: endocrine disruptors as carcinogens. Nat Rev Endocrinol 6:363–370 [PMC free article] [PubMed] [Google Scholar]
15. Meeker JD. 2010. Exposure to environmental endocrine disrupting compounds and men’s health. Maturitas 66:236–241 [PubMed] [Google Scholar]
16. Hatch EE, Nelson JW, Stahlhut RW, Webster TF. 2010. Association of endocrine disruptors and obesity: perspectives from epidemiological studies. Int J Androl 33:324–332 [PMC free article] [PubMed] [Google Scholar]
17. Hsu ST, Ma CI, Hsu SK, Wu SS, Hsu NH, Yeh CC, Wu SB. 1985. Discovery and epidemiology of PCB poisoning in Taiwan: a four-year followup. Environ Health Perspect 59:5–10 [PMC free article] [PubMed] [Google Scholar]
18. Pesatori AC, Consonni D, Bachetti S, Zocchetti C, Bonzini M, Baccarelli A, Bertazzi PA. 2003. Short- and long-term morbidity and mortality in the population exposed to dioxin after the “Seveso accident”. Ind Health 41:127–138 [PubMed] [Google Scholar]
19. Anderson HA, Wolff MS, Lilis R, Holstein EC, Valciukas JA, Anderson KE, Petrocci M, Sarkozi L, Selikoff IJ. 1979. Symptoms and clinical abnormalities following ingestion of polyborminated-biphenyl-contaminated food products. Ann NY Acad Sci 320:684–702 [PubMed] [Google Scholar]
20. Villeneuve S, Cyr D, Lynge E, Orsi L, Sabroe S, Merletti F, Gorini G, Morales-Suarez-Varela M, Ahrens W, Baumgardt-Elms C, Kaerlev L, Eriksson M, Hardell L, Févotte J, Guénel P. 2010. Occupation and occupational exposure to endocrine disrupting chemicals in male breast cancer: a case-control study in Europe. Occup Environ Med 67:837–844 [PMC free article] [PubMed] [Google Scholar]
21. Li D, Zhou Z, Qing D, He Y, Wu T, Miao M, Wang J, Weng X, Ferber JR, Herrinton LJ, Zhu Q, Gao E, Checkoway H, Yuan W. 2010. Occupational exposure to bisphenol-A (BPA) and the risk of self-reported male sexual dysfunction. Hum Reprod 25:519–527 [PubMed] [Google Scholar]
22. Queiroz EK, Waissmann W. 2006. Occupational exposure and effects on the male reproductive system. Cad Saude Publica 22:485–493 [PubMed] [Google Scholar]
23. Centers for Disease Control 2008. National Biomonitoring Program. Atlanta, GA: Centers for Disease Control, Prevention [Google Scholar]
24. Kuklenyik Z, Ye X, Needham LL, Calafat AM. 2009. Automated solid-phase extraction approaches for large scale biomonitoring studies. J Chromatogr Sci 47:12–18 [PubMed] [Google Scholar]
25. Umweltbundesamt 2009. Health and environmental hygiene: German environmental survey. Umweltbundesamt Dessau-Rosslau, Berlin, Germany [Google Scholar]
26. Ha MH, Lee DH, Son HK, Park SK, Jacobs DR., Jr 2009. Association between serum concentrations of persistent organic pollutants and prevalence of newly diagnosed hypertension: results from the National Health and Nutrition Examination Survey 1999–2002. J Hum Hypertens 23:274–286 [PubMed] [Google Scholar]
27. vom Saal FS, Akingbemi BT, Belcher SM, Birnbaum LS, Crain DA, Eriksen M, Farabollini F, Guillette LJ, Jr, Hauser R, Heindel JJ, Ho SM, Hunt PA, Iguchi T, Jobling S, Kanno J, Keri RA, Knudsen KE, Laufer H, LeBlanc GA, Marcus M, McLachlan JA, Myers JP, Nadal A, Newbold RR, Olea N, et al. 2007. Chapel Hill bisphenol A expert panel consensus statement: integration of mechanisms, effects in animals and potential to impact human health at current levels of exposure. Reprod Toxicol 24:131–138 [PMC free article] [PubMed] [Google Scholar]
28. Crain DA, Eriksen M, Iguchi T, Jobling S, Laufer H, LeBlanc GA, Guillette LJ., Jr 2007. An ecological assessment of bisphenol-A: evidence from comparative biology. Reprod Toxicol 24:225–239 [PubMed] [Google Scholar]
29. Richter CA, Birnbaum LS, Farabollini F, Newbold RR, Rubin BS, Talsness CE, Vandenbergh JG, Walser-Kuntz DR, vom Saal FS. 2007. In vivo effects of bisphenol A in laboratory rodent studies. Reprod Toxicol 24:199–224 [PMC free article] [PubMed] [Google Scholar]
30. Wetherill YB, Akingbemi BT, Kanno J, McLachlan JA, Nadal A, Sonnenschein C, Watson CS, Zoeller RT, Belcher SM. 2007. In vitro molecular mechanisms of bisphenol A action. Reprod Toxicol 24:178–198 [PubMed] [Google Scholar]31. Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. 2009. Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocrine Reviews 30:75–95 [PMC free article] [PubMed] [Google Scholar]32. Keri RA, Ho SM, Hunt PA, Knudsen KE, Soto AM, Prins GS. 2007. An evaluation of evidence for the carcinogenic activity of bisphenol A. Reprod Toxicol 24:240–252 [PMC free article] [PubMed] [Google Scholar]33. U.S. Food and Drug Administration 2008. Draft assessment of bisphenol A for use in food contact applications. Washington, DC: Department of Health and Human Services [Google Scholar]34. U.S. Food and Drug Administration 2010. Update on bisphenol A (BPA) for use in food: January 2010. Washington, DC: Department of Health and Human Services [Google Scholar]35. Soto AM, Sonnenschein C, Chung KL, Fernandez MF, Olea N, Serrano FO. 1995. The E-SCREEN assay as a tool to identify estrogens: an update on estrogenic environmental pollutants. Environ Health Perspect 103(Suppl 7):113–122 [PMC free article] [PubMed] [Google Scholar]36. Nagel SC, vom Saal FS, Welshons WV. 1999. Developmental effects of estrogenic chemicals are predicted by an in vitro assay incorporating modification of cell uptake by serum. J Steroid Biochem Mol Biol 69:343–357 [PubMed] [Google Scholar]37. Soto AM, Chung KL, Sonnenschein C. 1994. The pesticides endosulfan, toxaphene, and dieldrin have estrogenic effects on human estrogen-sensitive cells. Environ Health Perspect 102:380–383 [PMC free article] [PubMed] [Google Scholar]38. Welshons WV, Thayer KA, Judy BM, Taylor JA, Curran EM, vom Saal FS. 2003. Large effects from small exposures: I. Mechanisms for endocrine-disrupting chemicals with estrogenic activity. Environ Health Perspect 111:994–1006 [PMC free article] [PubMed] [Google Scholar]39. Kochukov MY, Jeng YJ, Watson CS. 2009. Alkylphenol xenoestrogens with varying carbon chain lengths differentially and potently activate signaling and functional responses in GH3/B6/F10 somatomammotropes. Environ Health Perspect 117:723–730 [PMC free article] [PubMed] [Google Scholar]40. Alyea RA, Watson CS. 2009. Differential regulation of dopamine transporter function and location by low concentrations of environmental estrogens and 17β-estradiol. Environ Health Perspect 117:778–783 [PMC free article] [PubMed] [Google Scholar]41. Wozniak AL, Bulayeva NN, Watson CS. 2005. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-α mediated Ca2+ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Environ Health Perspect 113:431–439 [PMC free article] [PubMed] [Google Scholar]42. Kohn MC, Melnick RL. 2002. Biochemical origins of the non-monotonic receptor-mediated dose-response. J Mol Endocrinol 29:113–123 [PubMed] [Google Scholar]43. Conolly RB, Lutz WK. 2004. Nonmonotonic dose-response relationships: mechanistic basis, kinetic modeling, and implications for risk assessment. Toxicol Sci 77:151–157 [PubMed] [Google Scholar]44. Zsarnovszky A, Le HH, Wang HS, Belcher SM. 2005. Ontogeny of rapid estrogen-mediated extracellular signal-regulated kinase signaling in the rat cerebellar cortex: potent nongenomic agonist and endocrine disrupting activity of the xenoestrogen bisphenol A. Endocrinology 146:5388–5396 [PubMed] [Google Scholar]45. Wong JK, Le HH, Zsarnovszky A, Belcher SM. 2003. Estrogens and ICI182,780 (Faslodex) modulate mitosis and cell death in immature cerebellar neurons via rapid activation of p44/p42 mitogen-activated protein kinase. J Neurosci 23:4984–4995 [PMC free article] [PubMed] [Google Scholar]46. Querfeld U, Mak RH. 2010. Vitamin D deficiency and toxicity in chronic kidney disease: in search of the therapeutic window. Pediatr Nephrol 25:2413–2430 [PubMed] [Google Scholar]47. Cook R, Calabrese EJ. 2006. The importance of hormesis to public health. Environ Health Perspect 114:1631–1635 [PMC free article] [PubMed] [Google Scholar]48. Thayer KA, Melnick R, Huff J, Burns K, Davis D. 2006. Hormesis: a new religion? Environ Health Perspect 114:A632–A633 [PMC free article] [PubMed] [Google Scholar]49. Weltje L, vom Saal FS, Oehlmann J. 2005. Reproductive stimulation by low doses of xenoestrogens contrasts with the view of hormesis as an adaptive response. Hum Exp Toxicol 24:431–437 [PubMed] [Google Scholar]50. Thayer KA, Melnick R, Burns K, Davis D, Huff J. 2005. Fundamental flaws of hormesis for public health decisions. Environ Health Perspect 113:1271–1276 [PMC free article] [PubMed] [Google Scholar]51. Beronius A, Rudén C, Håkansson H, Hanberg A. 2010. Risk to all or none? A comparative analysis of controversies in the health risk assessment of bisphenol A. Reprod Toxicol 29:132–146 [PubMed] [Google Scholar]52. Bellinger DC. 2004. What is an adverse effect? A possible resolution of clinical and epidemiological perspectives on neurobehavioral toxicity. Environ Res 95:394–405 [PubMed] [Google Scholar]53. Foster PM, McIntyre BS. 2002. Endocrine active agents: implications of adverse and non-adverse changes. Toxicol Pathol 30:59–65 [PubMed] [Google Scholar]54. Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, Calafat AM, Mao CS, Redmon JB, Ternand CL, Sullivan S, Teague JL. 2005. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect 113:1056–1061 [PMC free article] [PubMed] [Google Scholar]55. McEwen GN, Jr, Renner G. 2006. Validity of anogenital distance as a marker of in utero phthalate exposure. Environ Health Perspect 114:A19–A20; author reply A20–A21 [PMC free article] [PubMed] [Google Scholar]56. Weiss B. 2006. Anogenital distance: defining “normal.” Environ Health Perspect 114:A399; author reply A399 [PMC free article] [PubMed] [Google Scholar]57. Witorsch RJ. 2002. Low-dose in utero effects of xenoestrogens in mice and their relevance to humans: an analytical review of the literature. Food Chem Toxicol 40:905–912 [PubMed] [Google Scholar]58. O’Lone R, Frith MC, Karlsson EK, Hansen U. 2004. Genomic targets of nuclear estrogen receptors. Mol Endocrinol 18:1859–1875 [PubMed] [Google Scholar]59. Schulkin J. 2011. Evolutionary conservation of glucocorticoids and corticotropin releasing hormone: behavioral and physiological adaptations. Brain Res 1392:27–46 [PubMed] [Google Scholar]60. Williams GR, Franklyn JA. 1994. Physiology of the steroid-thyroid hormone nuclear receptor superfamily. Baillieres Clin Endocrinol Metab 8:241–266 [PubMed] [Google Scholar]61. Enmark E, Gustafsson JA. 1999. Oestrogen receptors: an overview. J Intern Med 246:133–138 [PubMed] [Google Scholar]62. U.S. Food and Drug Administration 2009. Information for consumers (drugs). In: The beginnings: laboratory and animal studies. Washington, DC: Department of Health and Human Services [Google Scholar]63. Mittendorf R. 1995. Teratogen update: carcinogenesis and teratogenesis associated with exposure to diethylstilbestrol (DES) in utero. Teratology 51:435–445 [PubMed] [Google Scholar]64. McLachlan JA. 2006. Commentary: prenatal exposure to diethylstilbestrol (DES): a continuing story. Int J Epidemiol 35:868–870 [PubMed] [Google Scholar]65. Newbold RR, Jefferson WN, Padilla-Banks E. 2007. Long-term adverse effects of neonatal exposure to bisphenol A on the murine female reproductive tract. Reprod Toxicol 24:253–258 [PMC free article] [PubMed] [Google Scholar]66. Palmer JR, Wise LA, Hatch EE, Troisi R, Titus-Ernstoff L, Strohsnitter W, Kaufman R, Herbst AL, Noller KL, Hyer M, Hoover RN. 2006. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev 15:1509–1514 [PubMed] [Google Scholar]67. Soto AM, Vandenberg LN, Maffini MV, Sonnenschein C. 2008. Does breast cancer start in the womb? Basic Clin Pharmacol Toxicol 102:125–133 [PMC free article] [PubMed] [Google Scholar]68. Kamrin MA. 2007. The “low dose” hypothesis: validity and implications for human risk. Int J Toxicol 26:13–23 [PubMed] [Google Scholar]69. Myers JP, vom Saal FS, Akingbemi BT, Arizono K, Belcher S, Colborn T, Chahoud I, Crain DA, Farabollini F, Guillette LJ, Jr, Hassold T, Ho SM, Hunt PA, Iguchi T, Jobling S, Kanno J, Laufer H, Marcus M, McLachlan JA, Nadal A, Oehlmann J, Olea N, Palanza P, Parmigiani S, Rubin BS, et al. 2009. Why public health agencies cannot depend upon ‘Good Laboratory Practices’ as a criterion for selecting data: the case of bisphenol-A. Environ Health Perspect 117:309–315 [PMC free article] [PubMed] [Google Scholar]70. Myers JP, Zoeller RT, vom Saal FS. 2009. A clash of old and new scientific concepts in toxicity, with important implications for public health. Environ Health Perspect 117:1652–1655 [PMC free article] [PubMed] [Google Scholar]71. vom Saal FS, Akingbemi BT, Belcher SM, Crain DA, Crews D, Guidice LC, Hunt PA, Leranth C, Myers JP, Nadal A, Olea N, Padmanabhan V, Rosenfeld CS, Schneyer A, Schoenfelder G, Sonnenschein C, Soto AM, Stahlhut RW, Swan SH, Vandenberg LN, Wang HS, Watson CS, Welshons WV, Zoeller RT. 2010. Flawed experimental design reveals the need for guidelines requiring appropriate positive controls in endocrine disruption research. Toxicol Sci 115:612–613; author reply 614–620 [PubMed] [Google Scholar]72. vom Saal FS, Myers JP. 2010. Good laboratory practices are not synonymous with good scientific practices, accurate reporting, or valid data. Environ Health Perspect 118:A60. [PMC free article] [PubMed] [Google Scholar]73. Travis GD. 1981. Replicating replication? Aspects of the social construction of learning in planarian worms. Social Studies Sci 11:11–32 [Google Scholar]74. Phillips CV, Goodman KJ. 2004. The missed lessons of Sir Austin Bradford Hill. Epidemiol Pespect Innov 1:3 [PMC free article] [PubMed] [Google Scholar]75. vom Saal FS, Hughes C. 2005. An extensive new literature concerning low-dose effects of bisphenol A shows the need for a new risk assessment. Environ Health Perspect 113:926–933 [PMC free article] [PubMed] [Google Scholar]76. Hayes TB. 2004. There is no denying this: defusing the confusion about atrazine. BioScience 54:1138–1149 [Google Scholar]77. vom Saal FS, Welshons WV. 2006. Large effects from small exposures. II. The importance of positive controls in low-dose research on bisphenol A. Environmental Research 100:50–76 [PubMed] [Google Scholar]78. Bern HA, Edery M, Mills KT, Kohrman AF, Mori T, Larson L. 1987. Long-term alterations in histology and steroid receptor levels of the genital tract and mammary gland following neonatal exposure of female BALB/cCrgl mice to various doses of diethylstilbestrol. Cancer Res 47:4165–4172 [PubMed] [Google Scholar]79. Krimsky S. 2003. Hormonal chaos: the scientific and social origins of the environmental endocrine hypothesis. Baltimore: Johns Hopkins University Press [Google Scholar]80. Barker DJ. 2007. The origins of the developmental origins theory. J Intern Med 261:412–417 [PubMed] [Google Scholar]81. Barker DJP. 2004. The developmental origins of adult disease. J Am Coll Nutr 23:588S–595S [PubMed] [Google Scholar]82. Sharpe RM, Skakkebaek NE. 1993. Are oestrogens involved in falling sperm counts and disorders of the male reproductive tract? Lancet 341:1392–1395 [PubMed] [Google Scholar]83. Trichopoulos D. 1990. Is breast cancer inititated in utero? Epidemiology 1:95–96 [PubMed] [Google Scholar]84. Heindel JJ. 2006. Role of exposure to environmental chemicals in the developmental basis of reproductive disease and dysfunction. Semin Reprod Med 24:168–177 [PubMed] [Google Scholar]85. Crain DA, Janssen SJ, Edwards TM, Heindel J, Ho SM, Hunt P, Iguchi T, Juul A, McLachlan JA, Schwartz J, Skakkebaek N, Soto AM, Swan S, Walker C, Woodruff TK, Woodruff TJ, Giudice LC, Guillette LJ., Jr 2008. Female reproductive disorders: the roles of endocrine-disrupting compounds and developmental timing. Fertil Steril 90:911–940 [PMC free article] [PubMed] [Google Scholar]86. Heindel JJ. 2005. The fetal basis of adult disease: Role of environmental exposures: introduction. Birth Defects Res A Clin Mol Teratol 73:131–132 [PubMed] [Google Scholar]87. Vandenberg LN, Chahoud I, Heindel JJ, Padmanabhan V, Paumgartten FJ, Schoenfelder G. 2010. Urine, serum and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environ Health Perspect 118:1055–1070 [PMC free article] [PubMed] [Google Scholar]88. Hays SM, Aylward LL. 2009. Using biomonitoring equivalents to interpret human biomonitoring data in a public health risk context. J Appl Toxicol 29:275–288 [PubMed] [Google Scholar]89. Clewell HJ, Tan YM, Campbell JL, Andersen ME. 2008. Quantitative interpretation of human biomonitoring data. Toxicol Appl Pharmacol 231:122–133 [PubMed] [Google Scholar]90. Hayes TB, Case P, Chui S, Chung D, Haeffele C, Haston K, Lee M, Mai VP, Marjuoa Y, Parker J, Tsui M. 2006. Pesticide mixtures, endocrine disruption, and amphibian declines: are we underestimating the impact? Environ Health Perspect 114:40–50 [PMC free article] [PubMed] [Google Scholar]91. Woodruff TJ, Zota AR, Schwartz JM. 2011. Environmental chemicals in pregnant women in the US: NHANES 2003–2004. Environ Health Perspect 119:878–885 [PMC free article] [PubMed] [Google Scholar]92. Young SS, Yu M. 2009. Association of bisphenol A with diabetes and other abnormalities. JAMA 301:720–721 [PubMed] [Google Scholar]93. Smith GD, Ebrahim S. 2002. Data dredging, bias, or confounding. BMJ 325:1437–1438 [PMC free article] [PubMed] [Google Scholar]94. Marshall JR. 1990. Data dredging and noteworthiness. Epidemiology 1:5–7 [PubMed] [Google Scholar]95. Vandenbroucke JP. 2008. Observational research, randomised trials, and two views of medical science. PLoS Medicine 5:e67. [PMC free article] [PubMed] [Google Scholar]96. Greenland S. 2007. Commentary: on ‘quality in epidemiological research: should we be submitting papers before we have the results and submitting more hypothesis generating research?’. Int J Epidemiol 36:944–945 [PubMed] [Google Scholar]97. Melzer D, Lang IA, Galloway TS. 2009. Reply to Young and Yu: association of bisphenol A with diabetes and other abnormalities. JAMA 301:721–722 [PubMed] [Google Scholar]98. Wigle DT, Arbuckle TE, Turner MC, Bérubé A, Yang Q, Liu S, Krewski D. 2008. Epidemiologic evidence of relationships between reproductive and child health outcomes and environmental chemical contaminants. J Toxicol Environ Health B Crit Rev 11:373–517 [PubMed] [Google Scholar]99. Watson CS, Gametchu B. 1999. Membrane-initiated steroid actions and the proteins that mediate them. Proc Soc Exp Biol Med 220:9–19 [PubMed] [Google Scholar]100. Frühbeck G. 2006. Intracellular signalling pathways activated by leptin. Biochem J 393:7–20 [PMC free article] [PubMed] [Google Scholar]101. George JW, Dille EA, Heckert LL. 2011. Current concepts of follicle-stimulating hormone receptor gene regulation. Biol Reprod 84:7–17 [PMC free article] [PubMed] [Google Scholar]102. Cheng SY, Leonard JL, Davis PJ. 2010. Molecular aspects of thyroid hormone actions. Endocr Rev 31:139–170 [PMC free article] [PubMed] [Google Scholar]103. Kress E, Samarut J, Plateroti M. 2009. Thyroid hormones and the control of cell proliferation or cell differentiation: paradox or duality? Mol Cell Endocrinol 313:36–49 [PubMed] [Google Scholar]104. Fu M, Wang C, Zhang X, Pestell RG. 2004. Acetylation of nuclear receptors in cellular growth and apoptosis. Biochem Pharmacol 68:1199–1208 [PubMed] [Google Scholar]105. Katzenellenbogen BS, Montano MM, Ediger TR, Sun J, Ekena K, Lazennec G, Martini PG, McInerney EM, Delage-Mourroux R, Weis K, Katzenellenbogen JA. 2000. Estrogen receptors: selective ligands, partners, and distinctive pharmacology. Recent Prog Horm Res 55:163–193; discussion 194–195 [PubMed] [Google Scholar]106. Zhao C, Dahlman-Wright K, Gustafsson JA. 2008. Estrogen receptor β: an overview and update. Nucl Recept Signal 6:e003. [PMC free article] [PubMed] [Google Scholar]107. Neill JD. 2005. Knobil and Neill’s physiology of reproduction. 3rd ed New York: Academic Press [Google Scholar]108. Jones KA. 1996. Summation of basic endocrine data. In: Gass GH, Kaplan HM, eds. Handbook of endocrinology. 2nd ed New York: CRC Press; 1–42 [Google Scholar]109. Stokes WS. 2004. Selecting appropriate animal models and experimental designs for endocrine disruptor research and testing studies. ILAR J 45:387–393 [PubMed] [Google Scholar]110. May M, Moran JF, Kimelberg H, Triggle DJ. 1967. Studies on the noradrenaline α-receptor. II. Analysis of the “spare-receptor” hypothesis and estimation of the concentration of α-receptors in rabbit aorta. Mol Pharmacol 3:28–36 [PubMed] [Google Scholar]111. Zhu BT. 1996. Rational design of receptor partial aganists and possible mechanisms of receptor partial activation: a theory. J Theor Biol 181:273–291 [PubMed] [Google Scholar]112. Gan EH, Quinton R. 2010. Physiological significance of the rhythmic secretion of hypothalamic and pituitary hormones. Prog Brain Res 181:111–126 [PubMed] [Google Scholar]113. Naftolin F, Garcia-Segura LM, Horvath TL, Zsarnovszky A, Demir N, Fadiel A, Leranth C, Vondracek-Klepper S, Lewis C, Chang A, Parducz A. 2007. Estrogen-induced hypothalamic synaptic plasticity and pituitary sensitization in the control of the estrogen-induced gonadotrophin surge. Reprod Sci 14:101–116 [PubMed] [Google Scholar]114. Son GH, Chung S, Kim K. 2011. The adrenal peripheral clock: glucocorticoid and the circadian timing system. Front Neuroendocrinol 32:451–465 [PubMed] [Google Scholar]115. Urbanski HF. 2011. Role of circadian neuroendocrine rhythms in the control of behavior and physiology. Neuroendocrinology 93:211–222 [PMC free article] [PubMed] [Google Scholar]116. National Research Council 1999. Hormonally active agents in the environment. Washington, DC: National Academy Press [Google Scholar]117. Eick GN, Thornton JW. 2011. Evolution of steroid receptors from an estrogen-sensitive ancestral receptor. Mol Cell Endocrinol 334:31–38 [PubMed] [Google Scholar]118. Sheehan DM. 2000. Activity of environmentally relevant low doses of endocrine disruptors and the bisphenol A controversy: initial results confirmed. Proc Soc Exp Biol Med 224:57–60 [PubMed] [Google Scholar]119. Hayes TB, Anderson LL, Beasley VR, de Solla SR, Iguchi T, Ingraham H, Kestemont P, Kniewald J, Kniewald Z, Langlois VS, Luque EH, McCoy KA, Muñoz-de-Toro M, Oka T, Oliveira CA, Orton F, Ruby S, Suzawa M, Tavera-Mendoza LE, Trudeau VL, Victor-Costa AB, Willingham E. 2011. Demasculinization and feminization of male gonads by atrazine: consistent effects across vertebrate classes. J Steroid Biochem Mol Biol 127:64–73 [PMC free article] [PubMed] [Google Scholar]120. Beato M, Klug J. 2000. Steroid hormone receptors: an update. Hum Reprod Update 6:225–236 [PubMed] [Google Scholar]121. Watson CS, Bulayeva NN, Wozniak AL, Finnerty CC. 2005. Signaling from the membrane via membrane estrogen receptor-α: estrogens, xenoestrogens, and phytoestrogens. Steroids 70:364–371 [PubMed] [Google Scholar]122. Powell CE, Soto AM, Sonnenschein C. 2001. Identification and characterization of membrane estrogen receptor from MCF7 estrogen-target cells. J Steroid Biochem Mol Biol 77:97–108 [PubMed] [Google Scholar]123. Levin ER. 2011. Extranuclear steroid receptors: roles in modulation of cell functions. Mol Endocrinol 25:377–384 [PMC free article] [PubMed] [Google Scholar]124. Levin ER. 2009. Plasma membrane estrogen receptors. Trends Endocrinol Metab 20:477–482 [PMC free article] [PubMed] [Google Scholar]125. Thomas P, Dong J. 2006. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol 102:175–179 [PubMed] [Google Scholar]126. Kenealy BP, Keen KL, Terasawa E. 2011. Rapid action of estradiol in primate GnRH neurons: The role of estrogen receptor α and estrogen receptor β. Steroids 76:861–866 [PMC free article] [PubMed] [Google Scholar]127. Watson CS, Bulayeva NN, Wozniak AL, Alyea RA. 2007. Xenoestrogens are potent activators of nongenomic estrogenic responses. Steroids 72:124–134 [PMC free article] [PubMed] [Google Scholar]128. Ropero AB, Alonso-Magdalena P, Ripoll C, Fuentes E, Nadal A. 2006. Rapid endocrine disruption: environmental estrogen actions triggered outside the nucleus. J Steroid Biochem Mol Biol 102:163–169 [PubMed] [Google Scholar]129. Nadal A, Alonso-Magdalena P, Ripoll C, Fuentes E. 2005. Disentangling the molecular mechanisms of action of endogenous and environmental estrogens. Pflugers Arch 449:335–343 [PubMed] [Google Scholar]130. Thomas P, Pang Y, Filardo EJ, Dong J. 2005. Identity of an estogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146:624–632 [PubMed] [Google Scholar]131. Nadal A, Ropero AB, Laribi O, Maillet M, Fuentes E, Soria B. 2000. Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor α and estrogen receptor β. Proc Natl Acad Sci USA 97:11603–11608 [PMC free article] [PubMed] [Google Scholar]132. Tanabe N, Kimoto T, Kawato S. 2006. Rapid Ca2+ signaling induced by bisphenol A in cultured rat hippocampal neurons. Neuro Endocrinol Lett 27:97–104 [PubMed] [Google Scholar]133. Ruehlmann DO, Steinert JR, Valverde MA, Jacob R, Mann GE. 1998. Environmental estrogenic pollutants induce acute vascular relaxation by inhibiting L-type Ca2+ channels in smooth muscle cells. FASEB J 12:613–619 [PubMed] [Google Scholar]134. Walsh DE, Dockery P, Doolan CM. 2005. Estrogen receptor independent rapid non-genomic effects of environmental estrogens on [Ca2+] in human breast cancer cells. Mol Cell Endocrinol 230:23–30 [PubMed] [Google Scholar]135. Shioda T, Chesnes J, Coser KR, Zou L, Hur J, Dean KL, Sonnenschein C, Soto AM, Isselbacher KJ. 2006. Importance of dosage standardization for interpreting transcriptomal signature profiles: evidence from studies of xenoestrogens. Proc Natl Acad Sci USA 103:12033–12038 [PMC free article] [PubMed] [Google Scholar]136. Ryan BC, Vandenbergh JG. 2002. Intrauterine position effects. Neurosci Biobehav Rev 26:665–678 [PubMed] [Google Scholar]137. Muñoz-de-Toro M, Markey CM, Wadia PR, Luque EH, Rubin BS, Sonnenschein C, Soto AM. 2005. Perinatal exposure to bisphenol-A alters peripubertal mammary gland development in mice. Endocrinology 146:4138–4147 [PMC free article] [PubMed] [Google Scholar]138. Wadia PR, Vandenberg LN, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. 2007. Perinatal bisphenol A exposure increases estrogen sensitivity of the mammary gland in diverse mouse strains. Environ Health Perspect 115:592–598 [PMC free article] [PubMed] [Google Scholar]139. Prins GS, Birch L, Tang WY, Ho SM. 2007. Developmental estrogen exposures predispose to prostate carcinogenesis with aging. Reprod Toxicol 23:374–382 [PMC free article] [PubMed] [Google Scholar]140. Prins GS, Tang WY, Belmonte J, Ho SM. 2008. Perinatal exposure to oestradiol and bisphenol A alters the prostate epigenome and increases susceptibility to carcinogenesis. Basic Clin Pharmacol Toxicol 102:134–138 [PMC free article] [PubMed] [Google Scholar]141. Prins GS, Ye SH, Birch L, Ho SM, Kannan K. 2011. Serum bisphenol A pharmacokinetics and prostate neoplastic responses following oral and subcutaneous exposures in neonatal Sprague-Dawley rats. Reprod Toxicol 31:1–9 [PMC free article] [PubMed] [Google Scholar]142. Bjørnerem A, Straume B, Midtby M, Fønnebø V, Sundsfjord J, Svartberg J, Acharya G, Oian P, Berntsen GK. 2004. Endogenous sex hormones in relation to age, sex, lifestyle factors, and chronic diseases in a general population: the Tromso Study. J Clin Endocrinol Metab 89:6039–6047 [PubMed] [Google Scholar]143. Silva E, Rajapakse N, Kortenkamp A. 2002. Something from “nothing”: eight weak estrogenic chemicals combined at concentrations below NOECs produce significant mixture effects. Environ Sci Technol 36:1751–1756 [PubMed] [Google Scholar]144. Soto AM, Fernandez MF, Luizzi MF, Oles Karasko AS, Sonnenschein C. 1997. Developing a marker of exposure to xenoestrogen mixtures in human serum. Environ Health Perspect 105:647–654 [PMC free article] [PubMed] [Google Scholar]145. Crofton KM. 2008. Thyroid disrupting chemicals: mechanisms and mixtures. Int J Androl 31:209–223 [PubMed] [Google Scholar]146. Montano MM, Welshons WV, vom Saal FS. 1995. Free estradiol in serum and brain uptake of estradiol during fetal and neonatal sexual differentiation in female rats. Biol Reprod 53:1198–1207 [PubMed] [Google Scholar]147. Nunez EA, Benassayag C, Savu L, Vallette G, Delorme J. 1979. Oestrogen binding function of α 1-fetoprotein. J Steroid Biochem 11:237–243 [PubMed] [Google Scholar]148. Milligan SR, Khan O, Nash M. 1998. Competitive binding of xenobiotic oestrogens to rat α-fetoprotein and to sex steroid binding proteins in human and rainbow trout (Oncorhynchus mykiss) plasma. Gen Comp Endocrinol 112:89–95 [PubMed] [Google Scholar]149. Sheehan DM, Young M. 1979. Diethylstilbestrol and estradiol binding to serum albumin and pregnancy plasma of rat and human. Endocrinology 104:1442–1446 [PubMed] [Google Scholar]150. Déchaud H, Ravard C, Claustrat F, de la Perrière AB, Pugeat M. 1999. Xenoestrogen interaction with human sex hormone-binding globulin (hSHBG). Steroids 64:328–334 [PubMed] [Google Scholar]151. Liu SV, Schally AV, Hawes D, Xiong S, Fazli L, Gleave M, Cai J, Groshen S, Brands F, Engel J, Pinski J. 2010. Expression of receptors for luteinizing hormone-releasing hormone (LH-RH) in prostate cancers following therapy with LH-RH agonists. Clin Cancer Res 16:4675–4680 [PubMed] [Google Scholar]152. Piccart M, Parker LM, Pritchard KI. 2003. Oestrogen receptor downregulation: an opportunity for extending the window of endocrine therapy in advanced breast cancer. Ann Oncol 14:1017–1025 [PubMed] [Google Scholar]153. Grandien K, Berkenstam A, Gustafsson JA. 1997. The estrogen receptor gene: promoter organization and expression. Int J Biochem Cell Biol 29:1343–1369 [PubMed] [Google Scholar]154. Morani A, Warner M, Gustafsson JA. 2008. Biological functions and clinical implications of oestrogen receptors alfa and β in epithelial tissues. J Intern Med 264:128–142 [PubMed] [Google Scholar]155. Mostaghel EA, Montgomery RB, Lin DW. 2007. The basic biochemistry and molecular events of hormone therapy. Curr Urol Rep 8:224–232 [PubMed] [Google Scholar]156. Phoenix CH, Goy RW, Gerall AA, Young WC. 1959. Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology 65:369–382 [PubMed] [Google Scholar]157. Vom Saal FS, Moyer CL. 1985. Prenatal effects on reproductive capacity during aging in female mice. Biol Reprod 32:1116–1126 [PubMed] [Google Scholar]158. Alonso-Magdalena P, Vieira E, Soriano S, Menes L, Burks D, Quesada I, Nadal A. 2010. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect 118:1243–1250 [PMC free article] [PubMed] [Google Scholar]159. Even MD, Dhar MG, vom Saal FS. 1992. Transport of steroids between fetuses via amniotic fluid in relation to the intrauterine position phenomenon in rats. J Reprod Fertil 96:709–716 [PubMed] [Google Scholar]160. vom Saal FS, Quadagno DM, Even MD, Keisler LW, Keisler DH, Khan S. 1990. Paradoxical effects of maternal stress on fetal steroids and postnatal reproductive traits in female mice from different intrauterine positions. Biol Reprod 43:751–761 [PubMed] [Google Scholar]161. vom Saal FS, Bronson FH. 1978. In utero proximity of female mouse fetuses to males: effect on reproductive performance during later life. Biol Reprod 19:842–853 [PubMed] [Google Scholar]162. Kinsley CH, Konen CM, Miele JL, Ghiraldi L, Svare B. 1986. Intrauterine position modulates maternal behaviors in female mice. Physiol Behav 36:793–799 [PubMed] [Google Scholar]163. Gandelman R, vom Saal FS, Reinisch JM. 1977. Contiguity to male foetuses affects morphology and behaviour of female mice. Nature 266:722–724 [PubMed] [Google Scholar]164. Palanza P, Parmigiani S, vom Saal FS. 1995. Urine marking and maternal aggression of wild female mice in relation to anogenital distance at birth. Physiol Behav 58:827–835 [PubMed] [Google Scholar]165. vom Saal FS, Grant WM, McMullen CW, Laves KS. 1983. High fetal estrogen concentrations: correlation with increased adult sexual activity and decreased aggression in male mice. Science 220:1306–1309 [PubMed] [Google Scholar]166. Palanza P, Morley-Fletcher S, Laviola G. 2001. Novelty seeking in periadolescent mice: sex differences and influence of intrauterine position. Physiol Behav 72:255–262 [PubMed] [Google Scholar]167. Clark MM, vom Saal FS, Galef BG., Jr 1992. Intrauterine positions and testosterone levels of adult male gerbils are correlated. Physiol Behav 51:957–960 [PubMed] [Google Scholar]168. vom Saal FS. 1989. Sexual differentiation in litter-bearing mammals: influence of sex of adjacent fetuses in utero. J Anim Sci 67:1824–1840 [PubMed] [Google Scholar]169. vom Saal FS. 1989. The production of and sensitivity to cues that delay puberty and prolong subsequent oestrous cycles in female mice are influenced by prior intrauterine position. J Reprod Fertil 86:457–471 [PubMed] [Google Scholar]170. Vom Saal FS, Even MD, Quadagno DM. 1991. Effects of maternal stress on puberty, fertility and aggressive behavior of female mice from different intrauterine positions. Physiol Behav 49:1073–1078 [PubMed] [Google Scholar]171. vom Saal FS, Pryor S, Bronson FH. 1981. Effects of prior intrauterine position and housing on oestrous cycle length in adolescent mice. Journal of Reproduction, Fertility 62:33–37 [PubMed] [Google Scholar]172. Vandenbergh JG, Huggett CL. 1994. Mother’s prior intrauterine position affects the sex ratio of her offspring in house mice. Proc Natl Acad Sci USA 91:11055–11059 [PMC free article] [PubMed] [Google Scholar]173. Vandenbergh JG, Huggett CL. 1995. The anogenital distance index, a predictor of the intrauterine position effects on reproduction in female house mice. Lab Anim Sci 45:567–573 [PubMed] [Google Scholar]174. Howdeshell KL, Hotchkiss AK, Thayer KA, Vandenbergh JG, vom Saal FS. 1999. Exposure to bisphenol A advances puberty. Nature 401:763–764 [PubMed] [Google Scholar]175. vom Saal FS, Bronson FH. 1980. Variation in length of estrous cycles in mice due to former intrauterine proximity to male fetuses. Biol Reprod 22:777–780 [PubMed] [Google Scholar]176. Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. 2007. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-A alters development of the fetal mouse mammary gland. Endocrinology 148:116–127 [PMC free article] [PubMed] [Google Scholar]177. Timms BG, Petersen SL, vom Saal FS. 1999. Prostate gland growth during development is stimulated in both male and female rat fetuses by intrauterine proximity to female fetuses. J Urol 161:1694–1701 [PubMed] [Google Scholar]178. Nonneman DJ, Ganjam VK, Welshons WV, Vom Saal FS. 1992. Intrauterine position effects on steroid metabolism and steroid receptors of reproductive organs in male mice. Biol Reprod 47:723–729 [PubMed] [Google Scholar]179. Clark MM, Bishop AM, vom Saal FS, Galef BG., Jr 1993. Responsiveness to testosterone of male gerbils from known intrauterine positions. Physiol Behav 53:1183–1187 [PubMed] [Google Scholar]180. vom Saal FS, Bronson FH. 1980. Sexual characteristics of adult female mice are correlated with their blood testosterone levels during prenatal development. Science 208:597–599 [PubMed] [Google Scholar]181. Timms BG, Peterson RE, vom Saal FS. 2002. 2,3,7,8-tetrachlorodibenzo-p-dioxin interacts with endogenous estradiol to disrupt prostate gland morphogenesis in male rat fetuses. Toxicol Sci 67:264–274 [PubMed] [Google Scholar]182. Vandenbergh JG. 2004. Animal models and studies of in utero endocrine disruptor effects. ILAR J 45:438–442 [PubMed] [Google Scholar]183. Clark MM, Crews D, Galef BG., Jr 1991. Concentrations of sex steroid hormones in pregnant and fetal Mongolian gerbils. Physiol Behav 49:239–243 [PubMed] [Google Scholar]184. Satoh S, Hirata T, Miyake Y, Kaneda Y. 1997. The possibility of early estimation for fertility in bovine heterosexual twin females. J Vet Med Sci 59:221–222 [PubMed] [Google Scholar]185. Padula AM. 2005. The freemartin syndrome: an update. Anim Reprod Sci 87:93–109 [PubMed] [Google Scholar]186. Resnick SM, Gottesman II, McGue M. 1993. Sensation seeking in opposite-sex twins: an effect of prenatal hormones? Behav Genet 23:323–329 [PubMed] [Google Scholar]187. McFadden D. 1993. A masculinizing effect on the auditory systems of human females having male co-twins. Proc Natl Acad Sci USA 90:11900–11904 [PMC free article] [PubMed] [Google Scholar]188. Cohen-Bendahan CC, Buitelaar JK, van Goozen SH, Cohen-Kettenis PT. 2004. Prenatal exposure to testosterone and functional cerebral lateralization: a study in same-sex and opposite-sex twin girls. Psychoneuroendocrinology 29:911–916 [PubMed] [Google Scholar]189. Peper JS, Brouwer RM, van Baal GC, Schnack HG, van Leeuwen M, Boomsma DI, Kahn RS, Hulshoff Pol HE. 2009. Does having a twin brother make for a bigger brain? Eur J Endocrinol 160:739–746 [PubMed] [Google Scholar]190. Cohen-Bendahan CC, Buitelaar JK, van Goozen SH, Orlebeke JF, Cohen-Kettenis PT. 2005. Is there an effect of prenatal testosterone on aggression and other behavioral traits? A study comparing same-sex and opposite-sex twin girls. Horm Behav 47:230–237 [PubMed] [Google Scholar]191. Loehlin JC, Martin NG. 2000. Dimensions of psychological masculinity-femininity in adult twins from opposite-sex and same-sex pairs. Behav Genet 30:19–28 [PubMed] [Google Scholar]192. Rose RJ, Kaprio J, Winter T, Dick DM, Viken RJ, Pulkkinen L, Koskenvuo M. 2002. Femininity and fertility in sisters with twin brothers: prenatal androgenization? Cross-sex socialization? Psychol Sci 13:263–267 [PubMed] [Google Scholar]193. Vuoksimaa E, Eriksson CJ, Pulkkinen L, Rose RJ, Kaprio J. 2010. Decreased prevalence of left-handedness among females with male co-twins: evidence suggesting prenatal testosterone transfer in humans? Psychoneuroendocrinology 35:1462–1472 [PMC free article] [PubMed] [Google Scholar]194. Elkadi S, Nicholls ME, Clode D. 1999. Handedness in opposite and same-sex dizygotic twins: testing the testosterone hypothesis. Neuroreport 10:333–336 [PubMed] [Google Scholar]195. Lummaa V, Pettay JE, Russell AF. 2007. Male twins reduce fitness of female co-twins in humans. Proc Natl Acad Sci USA 104:10915–10920 [PMC free article] [PubMed] [Google Scholar]196. van Anders SM, Vernon PA, Wilbur CJ. 2006. Finger-length ratios show evidence of prenatal hormone-transfer between opposite-sex twins. Horm Behav 49:315–319 [PubMed] [Google Scholar]197. Culbert KM, Breedlove SM, Burt SA, Klump KL. 2008. Prenatal hormone exposure and risk for eating disorders. Arch Gen Psychiatry 65:329–336 [PMC free article] [PubMed] [Google Scholar]198. Glinianaia SV, Magnus P, Harris JR, Tambs K. 1998. Is there a consequence for fetal growth of having an unlike-sexed cohabitant in utero? Int J Epidemiol 27:657–659 [PubMed] [Google Scholar]199. Cerhan JR, Kushi LH, Olson JE, Rich SS, Zheng W, Folsom AR, Sellers TA. 2000. Twinship and risk of postmenopausal breast cancer. J Natl Cancer Inst 92:261–265 [PubMed] [Google Scholar]200. Swerdlow AJ, De Stavola BL, Swanwick MA, Maconochie NES. 1997. Risks of breast and testicular cancers in young adult twins in England and Wales: evidence on prenatal and genetic aetiology. Lancet 350:1723–1728 [PubMed] [Google Scholar]201. van de Beek C, Thijssen JH, Cohen-Kettenis PT, van Goozen SH, Buitelaar JK. 2004. Relationships between sex hormones assessed in amniotic fluid, and maternal and umbilical cord serum: what is the best source of information to investigate the effects of fetal hormone exposure? Horm Behav 46:663–669 [PubMed] [Google Scholar]202. Sakai LM, Baker LA, Jacklin CN, Shulman I. 1991. Sex steroids at birth: genetic and environmental variation and covariation. Dev Psychobiol 24:559–570 [PubMed] [Google Scholar]203. Cohen-Bendahan CC, van Goozen SH, Buitelaar JK, Cohen-Kettenis PT. 2005. Maternal serum steroid levels are unrelated to fetal sex: a study in twin pregnancies. Twin Res Hum Genet 8:173–177 [PubMed] [Google Scholar]204. Johnson MR, Abbas A, Nicolaides KH. 1994. Maternal plasma levels of human chorionic gonadotropin, oestradiol and progesterone in multifetal pregnancies before and after fetal reduction. J Endocrinol 143:309–312 [PubMed] [Google Scholar]205. Vom Saal FS, Richter CA, Ruhlen RR, Nagel SC, Timms BG, Welshons WV. 2005. The importance of appropriate controls, animal feed, and animal models in interpreting results from low-dose studies of bisphenol A. Birth Defects Res A Clin Mol Teratol 73:140–145 [PubMed] [Google Scholar]206. Spearow JL, Doemeny P, Sera R, Leffler R, Barkley M. 1999. Genetic variation in susceptibility to endocrine disruption by estrogen in mice. Science 285:1259–1261 [PubMed] [Google Scholar]207. Spearow JL, O’Henley P, Doemeny P, Sera R, Leffler R, Sofos T, Barkley M. 2001. Genetic variation in physiological sensitivity to estrogen in mice. APMIS 109:356–364 [PubMed] [Google Scholar]208. Timms BG, Howdeshell KL, Barton L, Bradley S, Richter CA, vom Saal FS. 2005. Estrogenic chemicals in plastic and oral contraceptives disrupt development of the fetal mouse prostate and urethra. Proc Natl Acad Sci USA 102:7014–7019 [PMC free article] [PubMed] [Google Scholar]209. Cederroth CR, Nef S. 2009. Fetal programming of adult glucose homeostasis in mice. PLoS ONE 4:e7281. [PMC free article] [PubMed] [Google Scholar]210. Marty MS, Carney EW, Rowlands JC. 2011. Endocrine disruption: historical perspectives and its impact on the future of toxicology testing. Toxicol Sci 120:S93–S108 [PubMed] [Google Scholar]211. Bonefeld-Jorgensen EC, Long M, Hofmeister MV, Vinggaard AM. 2007. Endocrine-disrupting potential of bisphenol A, bisphenol A dimethacrylate, 4-n-nonylphenol, and 4-n-octylphenol in vitro: new data and a brief review. Environ Health Perspect 115(Suppl 1):69–76 [PMC free article] [PubMed] [Google Scholar]212. Krüger T, Long M, Bonefeld-Jørgensen EC. 2008. Plastic components affect the activation of the aryl hydrocarbon and the androgen receptor. Toxicology 246:112–123 [PubMed] [Google Scholar]213. Watson CS, Jeng YJ, Kochukov MY. 2010. Nongenomic signaling pathways of estrogen toxicity. Toxicol Sci 115:1–11 [PMC free article] [PubMed] [Google Scholar]214. Weed DL. 2005. Weight of evidence: a review of concepts and methods. Risk Anal 25:1545–1557 [PubMed] [Google Scholar]215. Linkov I, Loney D, Cormier S, Satterstrom FK, Bridges T. 2009. Weight-of-evidence evaluation in environmental assessment: review of qualitative and quantitative approaches. Sci Total Environ 407:5199–5205 [PubMed] [Google Scholar]216. Schreider J, Barrow C, Birchfield N, Dearfield K, Devlin D, Henry S, Kramer M, Schappelle S, Solomon K, Weed DL, Embry MR. 2010. Enhancing the credibility of decisions based on scientific conclusions: transparency is imperative. Toxicol Sci 116:5–7 [PubMed] [Google Scholar]217. Basketter D, Ball N, Cagen S, Carrillo JC, Certa H, Eigler D, Garcia C, Esch H, Graham C, Haux C, Kreiling R, Mehling A. 2009. Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Regul Toxicol Pharmacol 55:90–96 [PubMed] [Google Scholar]218. Wright-Walters M, Volz C, Talbott E, Davis D. 2011. An updated weight of evidence approach to the aquatic hazard assessment of bisphenol A and the derivation a new predicted no effect concentration (Pnec) using a non-parametric methodology. Sci Total Environ 409:676–685 [PubMed] [Google Scholar]219. Cooper RL, Kavlock RJ. 1997. Endocrine disruptors and reproductive development: a weight-of-evidence overview. J Endocrinol 152:159–166 [PubMed] [Google Scholar]220. Popp JA, Crouch E, McConnell EE. 2006. A weight-of-evidence analysis of the cancer dose-response characteristics of 2,3,7,8-tetrachlorodibenzodioxin (TCDD). Toxicol Sci 89:361–369 [PubMed] [Google Scholar]221. Goodman M, Squibb K, Youngstrom E, Anthony LG, Kenworthy L, Lipkin PH, Mattison DR, Lakind JS. 2010. Using systematic reviews and meta-analyses to support regulatory decision making for neurotoxicants: lessons learned from a case study of PCBs. Environ Health Perspect 118:727–734 [PMC free article] [PubMed] [Google Scholar]222. Goodman JE, Witorsch RJ, McConnell EE, Sipes IG, Slayton TM, Yu CJ, Franz AM, Rhomberg LR. 2009. Weight-of-evidence evaluation of reproductive and developmental effects of low doses of bisphenol A. Crit Rev Toxicol 39:1–75 [PubMed] [Google Scholar]223. Heindel JJ, vom Saal FS. 2008. Meeting report: batch-to-batch variability in estrogenic activity in commercial animal diets- importance and approaches for laboratory animal research. Environ Health Perspect 116:389–393 [PMC free article] [PubMed] [Google Scholar]224. Ruhlen RL, Taylor JA, Mao J, Kirkpatrick J, Welshons WV, vom Saal FS. 2011. Choice of animal feed can alter fetal steroid levels and mask developmental effects of endocrine disrupting chemicals. J Dev Origins Health Dis 2:36–48 [Google Scholar]225. vom Saal FS, Richter CA, Mao J, Welshons WV. 2005. Commercial animal feed: variability in estrogenic activity and effects on body weight in mice. Birth Defects Res (Part A) 73:474–475 [PubMed] [Google Scholar]226. Howdeshell KL, Peterman PH, Judy BM, Taylor JA, Orazio CE, Ruhlen RL, Vom Saal FS, Welshons WV. 2003. Bisphenol A is released from polycarbonate animal cages into water at room temperature. Environ Health Perspect 111:1180–1187 [PMC free article] [PubMed] [Google Scholar]227. Koehler KE, Voigt RC, Thomas S, Lamb B, Urban C, Hassold T, Hunt PA. 2003. When disaster strikes: rethinking caging materials. Lab Anim (NY) 32:24–27 [PubMed] [Google Scholar]228. Muhlhauser A, Susiarjo M, Rubio C, Griswold J, Gorence G, Hassold T, Hunt PA. 2009. Bisphenol A effects on the growing mouse oocyte are influenced by diet. Biol Reprod 80:1066–1071 [PMC free article] [PubMed] [Google Scholar]229. Tyl RW, Myers CB, Marr MC, Castillo NP, Veselica MM, Joiner RL, Dimond SS, Van Miller JP, Stropp GD, Waechter JM, Jr, Hentges SG. 2008. One-generation reproductive toxicity study of dietary 17β-estradiol (E2; CAS no. 50-28-2) in CD-1 (Swiss) mice. Reprod Toxicol 25:144–160 [PubMed] [Google Scholar]230. Ryan BC, Hotchkiss AK, Crofton KM, Gray LE., Jr 2010. In utero and lactational exposure to bisphenol A, in contrast to ethinyl estradiol, does not alter sexually dimorphic behavior, puberty, fertility, and anatomy of female LE rats. Toxicol Sci 114:133–148 [PubMed] [Google Scholar]231. Marty MS, Allen B, Chapin RE, Cooper R, Daston GP, Flaws JA, Foster PM, Makris SL, Mylchreest E, Sandler D, Tyl RW. 2009. Inter-laboratory control data for reproductive endpoints required in the OPPTS 870.3800/OECD 416 reproduction and fertility test. Birth Defects Res B Dev Reprod Toxicol 86:470–489 [PubMed] [Google Scholar]232. Teng CT, Beard C, Gladwell W. 2002. Differential expression and estrogen response of lactoferrin gene in the female reproductive tract of mouse, rat, and hamster. Biol Reprod 67:1439–1449 [PubMed] [Google Scholar]233. Aupperlee MD, Drolet AA, Durairaj S, Wang W, Schwartz RC, Haslam SZ. 2009. Strain-specific differences in the mechanisms of progesterone regulation of murine mammary gland development. Endocrinology 150:1485–1494 [PMC free article] [PubMed] [Google Scholar]234. Pepling ME, Sundman EA, Patterson NL, Gephardt GW, Medico L, Jr, Wilson KI. 2010. Differences in oocyte development and estradiol sensitivity among mouse strains. Reproduction 139:349–357 [PubMed] [Google Scholar]235. Wiklund JA, Gorski J. 1982. Genetic differences in estrogen-induced DNA synthesis in the rat pituitary: correlations with pituitary tumor susceptibility. Endocrinology 111:1140–1149 [PubMed] [Google Scholar]236. Wiklund J, Wertz N, Gorski J. 1981. A comparison of estrogen effects on uterine and pituitary growth and prolactin synthesis in F344 and Holtzman rats. Endocrinology 109:1700–1707 [PubMed] [Google Scholar]237. Diel P, Schmidt S, Vollmer G, Janning P, Upmeier A, Michna H, Bolt HM, Degen GH. 2004. Comparative responses of three rat strains (DA/Han, Sprague-Dawley and Wistar) to treatment with environmental estrogens. Arch Toxicol 78:183–193 [PubMed] [Google Scholar]238. Brossia LJ, Roberts CS, Lopez JT, Bigsby RM, Dynlacht JR. 2009. Interstrain differences in the development of pyometra after estrogen treatment of rats. J Am Assoc Lab Anim Sci 48:517–520 [PMC free article] [PubMed] [Google Scholar]239. Geis RB, Diel P, Degen GH, Vollmer G. 2005. Effects of genistein on the expression of hepatic genes in two rat strains (Sprague-Dawley and Wistar). Toxicol Lett 157:21–29 [PubMed] [Google Scholar]240. Roper RJ, Griffith JS, Lyttle CR, Doerge RW, McNabb AW, Broadbent RE, Teuscher C. 1999. Interacting quantitative trait loci control phenotypic variation in murine estradiol-regulated responses. Endocrinology 140:556–561 [PubMed] [Google Scholar]241. Taylor JA, Welshons WV, Vom Saal FS. 2008. No effect of route of exposure (oral; subcutaneous injection) on plasma bisphenol A throughout 24h after administration in neonatal female mice. Reprod Toxicol 25:169–176 [PMC free article] [PubMed] [Google Scholar]242. European Food Safety Authority 2007. Opinion of the Scientific Panel on food additives, flavourings, processing aids and materials in contact with food (AFC) related to 2,2-biS(4-hydroxyphenyl)propane. EFSA J 428:1–75 [Google Scholar]243. Vandenberg LN, Chahoud I, Padmanabhan V, Paumgartten FJ, Schoenfelder G. 2010. Biomonitoring studies should be used by regulatory agencies to assess human exposure levels and safety of bisphenol A. Environ Health Perspect 118:1051–1054 [PMC free article] [PubMed] [Google Scholar]244. Vandenberg LN. 2011. Exposure to bisphenol A in Canada: invoking the precautionary principle. CMAJ 183:1265–1270 [PMC free article] [PubMed] [Google Scholar]245. Stahlhut RW, Welshons WV, Swan SH. 2009. Bisphenol A data in NHANES suggest longer than expected half-life, substantial non-food exposure, or both. Environ Health Perspect 117:784–789 [PMC free article] [PubMed] [Google Scholar]246. Geens T, Goeyens L, Covaci A. 2011. Are potential sources for human exposure to bisphenol-A overlooked? Int J Hyg Environ Health 214:339–347 [PubMed] [Google Scholar]247. Biedermann S, Tschudin P, Grob K. 2010. Transfer of bisphenol A from thermal printer paper to the skin. Anal Bioanal Chem 398:571–576 [PubMed] [Google Scholar]248. Zalko D, Jacques C, Duplan H, Bruel S, Perdu E. 2011. Viable skin efficiently absorbs and metabolizes bisphenol A. Chemosphere 82:424–430 [PubMed] [Google Scholar]249. Moriyama K, Tagami T, Akamizu T, Usui T, Saijo M, Kanamoto N, Hataya Y, Shimatsu A, Kuzuya H, Nakao K. 2002. Thyroid hormone action is disrupted by bisphenol A as an antagonist. J Clin Endocrinol Metab 87:5185–5190 [PubMed] [Google Scholar]250. Zoeller RT, Bansal R, Parris C. 2005. Bisphenol-A, an environmental contaminant that acts as a thyroid hormone receptor antagonist in vitro, increases serum thyroxine, and alters RC3/neurogranin expression in the developing rat brain. Endocrinology 146:607–612 [PubMed] [Google Scholar]251. Lee HJ, Chattopadhyay S, Gong EY, Ahn RS, Lee K. 2003. Antiandrogenic effects of bisphenol A and nonphenol on the function of androgen receptor. Toxicol Sci 75:40–46 [PubMed] [Google Scholar]252. Kwintkiewicz J, Nishi Y, Yanase T, Giudice LC. 2010. Peroxisome proliferator-activated receptor-γ mediates bisphenol A inhibition of FSH-stimulated IGF-1, aromatase, and estradiol in human granulosa cells. Environ Health Perspect 118:400–406 [PMC free article] [PubMed] [Google Scholar]253. Taylor JA, Vom Saal FS, Welshons WV, Drury B, Rottinghaus G, Hunt PA, Toutain PL, Laffont CM, VandeVoort CA. 2011. Similarity of bisphenol A pharmacokinetics in rhesus monkeys and mice: relevance for human exposure. Environ Health Perspect 119:422–430 [PMC free article] [PubMed] [Google Scholar]254. Owens JW, Chaney JG. 2005. Weighing the results of differing ‘low dose’ studies of the mouse prostate by Nagel, Cagen, and Ashby: quantification of experimental power and statistical results. Regul Toxicol Pharmacol 43:194–202 [PubMed] [Google Scholar]255. Ashby J, Tinwell H, Odum J, Lefevre P. 2004. Natural variability and the influence of concurrent control values on the detection and interpretation of low-dose or weak endocrine toxicities. Environ Health Perspect 112:847–853 [PMC free article] [PubMed] [Google Scholar]256. Nagel SC, vom Saal FS, Thayer KA, Dhar MG, Boechler M, Welshons WV. 1997. Relative binding affinity-serum modified access (RBA-SMA) assay predicts the relative in vivo bioactivity of the xenoestrogens bisphenol A and octylphenol. Environ Health Perspect 105:70–76 [PMC free article] [PubMed] [Google Scholar]257. Gupta C. 2000. Reproductive malformation of the male offspring following maternal exposure to estrogenic chemicals. Proc Soc Exp Biol Med 224:61–68 [PubMed] [Google Scholar]258. Elswick BA, Welsch F, Janszen DB. 2000. Effect of different sampling designs on outcome of endocrine disruptor studies. Reprod Toxicol 14:359–367 [PubMed] [Google Scholar]259. Chitra KC, Latchoumycandane C, Mathur PP. 2003. Induction of oxidative stress by bisphenol A in the epididymal sperm of rats. Toxicology 185:119–127 [PubMed] [Google Scholar]260. Ramos JG, Varayoud J, Sonnenschein C, Soto AM, Muñoz De Toro M, Luque EH. 2001. Prenatal exposure to low doses of bisphenol A alters the periductal stroma and glandular cell function in the rat ventral prostate. Biol Reprod 65:1271–1277 [PubMed] [Google Scholar]261. Ramos JG, Varayoud J, Kass L, Rodríguez H, Costabel L, Muñoz-De-Toro M, Luque EH. 2003. Bisphenol A induces both transient and permanent histofunctional alterations of the hypothalamic-pituitary-gonadal axis in prenatally exposed male rats. Endocrinology 144:3206–3215 [PubMed] [Google Scholar]262. Ogura Y, Ishii K, Kanda H, Kanai M, Arima K, Wang Y, Sugimura Y. 2007. Bisphenol A induces permanent squamous change in mouse prostatic epithelium. Differentiation 75:745–756 [PubMed] [Google Scholar]263. Ho SM, Tang WY, Belmonte de Frausto J, Prins GS. 2006. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res 66:5624–5632 [PMC free article] [PubMed] [Google Scholar]264. Ichihara T, Yoshino H, Imai N, Tsutsumi T, Kawabe M, Tamano S, Inaguma S, Suzuki S, Shirai T. 2003. Lack of carcinogenic risk in the prostate with transplacental and lactational exposure to bisphenol A in rats. J Toxicol Sci 28:165–171 [PubMed] [Google Scholar]265. Ashby J, Tinwell H, Haseman J. 1999. Lack of effects for low dose levels of bisphenol A and diethylstilbestrol on the prostate gland of CF1 mice exposed in utero. Regul Toxicol Pharmacol 30:156–166 [PubMed] [Google Scholar]266. Cagen SZ, Waechter JM, Jr, Dimond SS, Breslin WJ, Butala JH, Jekat FW, Joiner RL, Shiotsuka RN, Veenstra GE, Harris LR. 1999. Normal reproductive organ development in CF-1 mice following prenatal exposure to bisphenol A. Toxicol Sci 50:36–44 [PubMed] [Google Scholar]267. Cagen SZ, Waechter JM, Jr, Dimond SS, Breslin WJ, Butala JH, Jekat FW, Joiner RL, Shiotsuka RN, Veenstra GE, Harris LR. 1999. Normal reproductive organ development in Wistar rats exposed to bisphenol A in the drinking water. Regul Toxicol Pharmacol 30:130–139 [PubMed] [Google Scholar]268. Ema M, Fujii S, Furukawa M, Kiguchi M, Ikka T, Harazono A. 2001. Rat two-generation reproductive toxicity study of bisphenol A. Reprod Toxicol 15:505–523 [PubMed] [Google Scholar]269. Tinwell H, Haseman J, Lefevre PA, Wallis N, Ashby J. 2002. Normal sexual development of two strains of rat exposed in utero to low doses of bisphenol A. Toxicol Sci 68:339–348 [PubMed] [Google Scholar]270. Tyl RW, Myers CB, Marr MC, Thomas BF, Keimowitz AR, Brine DR, Veselica MM, Fail PA, Chang TY, Seely JC, Joiner RL, Butala JH, Dimond SS, Cagen SZ, Shiotsuka RN, Stropp GD, Waechter JM. 2002. Three-generation reproductive toxicity study of dietary bisphenol A in CD Sprague-Dawley rats. Toxicol Sci 68:121–146 [PubMed] [Google Scholar]271. Tyl RW, Myers CB, Marr MC, Sloan CS, Castillo NP, Veselica MM, Seely JC, Dimond SS, Van Miller JP, Shiotsuka RN, Beyer D, Hentges SG, Waechter JM., Jr 2008. Two-generation reproductive toxicity study of dietary bisphenol A in CD-1 (Swiss) mice. Toxicol Sci 104:362–384 [PubMed] [Google Scholar]272. Howdeshell KL, Furr J, Lambright CR, Wilson VS, Ryan BC, Gray LE., Jr 2008. Gestational and lactational exposure to ethinyl estradiol, but not bisphenol A, decreases androgen-dependent reproductive organ weights and epididymal sperm abundance in the male long evans hooded rat. Toxicol Sci 102:371–382 [PubMed] [Google Scholar]273. Chapin RE, Adams J, Boekelheide K, Gray LE, Jr, Hayward SW, Lees PS, McIntyre BS, Portier KM, Schnorr TM, Selevan SG, Vandenbergh JG, Woskie SR. 2008. NTP-CERHR expert panel report on the reproductive and developmental toxicity of bisphenol A. Birth Defects Res B Dev Reprod Toxicol 83:157–395 [PubMed] [Google Scholar]274. Hennighausen L, Robinson GW. 1998. Think globally, act locally: the making of a mouse mammary gland. Genes Dev 12:449–455 [PubMed] [Google Scholar]275. Lemmen JG, Broekhof JL, Kuiper GG, Gustafsson JA, van der Saag PT, van der Burg B. 1999. Expression of estrogen receptor α and β during mouse embryogensis. Mech Dev 81:163–167 [PubMed] [Google Scholar]276. Padilla-Banks E, Jefferson WN, Newbold RR. 2006. Neonatal exposure to the phytoestrogen genistein alters mammary gland growth and developmental programming of hormone receptor levels. Endocrinology 147:4871–4882 [PubMed] [Google Scholar]277. Colerangle JB, Roy D. 1997. Profound effects of the weak environmental estrogen-like chemical bisphenol A on the growth of the mammary gland of Noble rats. J Steroid Biochem Mol Biol 60:153–160 [PubMed] [Google Scholar]278. Bern HA, Mills KT, Jones LA. 1983. Critical period of neonatal estrogen exposure in occurence of mammary gland abnormalities in adult mice. Proc Soc Exp Biol Med 172:239–242 [PubMed] [Google Scholar]279. Markey CM, Coombs MA, Sonnenschein C, Soto AM. 2003. Mammalian development in a changing environment: exposure to endocrine disruptors reveals the developmental plasticity of steroid-hormone target organs. Evol Dev 5:67–75 [PubMed] [Google Scholar]280. Markey CM, Luque EH, Munoz De Toro M, Sonnenschein C, Soto AM. 2001. In utero exposure to bisphenol A alters the development and tissue organization of the mouse mammary gland. Biol Reprod 65:1215–1223 [PubMed] [Google Scholar]281. Vandenberg LN, Maffini MV, Schaeberle CM, Ucci AA, Sonnenschein C, Rubin BS, Soto AM. 2008. Perinatal exposure to the xenoestrogen bisphenol-A induces mammary intraductal hyperplasias in adult CD-1 mice. Reprod Toxicol 26:210–219 [PMC free article] [PubMed] [Google Scholar]282. Moral R, Wang R, Russo IH, Lamartiniere CA, Pereira J, Russo J. 2008. Effect of prenatal exposure to the endocrine disruptor bisphenol A on mammary gland morphology and gene expression signature. J Endocrinol 196:101–112 [PubMed] [Google Scholar]283. Ayyanan A, Laribi O, Schuepbach-Mallepell S, Schrick C, Gutierrez M, Tanos T, Lefebvre G, Rougemont J, Yalcin-Ozuysal O, Brisken C. 2011. Perinatal exposure to bisphenol A increases adult mammary gland progesterone response and cell number. Mol Endocrinol 25:1915–1923 [PMC free article] [PubMed] [Google Scholar]284. Nikaido Y, Yoshizawa K, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, Tsubura A. 2004. Effects of maternal xenoestrogen exposure on development of the reproductive tract and mammary gland in female CD-1 mouse offspring. Reprod Toxicol 18:803–811 [PubMed] [Google Scholar]285. Jones LP, Sampson A, Kang HJ, Kim HJ, Yi YW, Kwon SY, Babus JK, Wang A, Bae I. 2010. Loss of BRCA1 leads to an increased sensitivity to bisphenol A. Toxicol Lett 199:261–268 [PMC free article] [PubMed] [Google Scholar]286. Murray TJ, Maffini MV, Ucci AA, Sonnenschein C, Soto AM. 2007. Induction of mammary gland ductal hyperplasias and carcinomas in situ following fetal bisphenol A exposure. Reprod Toxicol 23:383–390 [PMC free article] [PubMed] [Google Scholar]287. Durando M, Kass L, Piva J, Sonnenschein C, Soto AM, Luque EH, Muñoz-de-Toro M. 2007. Prenatal bisphenol A exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ Health Perspect 115:80–86 [PMC free article] [PubMed] [Google Scholar]288. Jenkins S, Raghuraman N, Eltoum I, Carpenter M, Russo J, Lamartiniere CA. 2009. Oral exposure to bisphenol A increases dimethylbenzanthracene-induced mammary cancer in rats. Environ Health Perspect 117:910–915 [PMC free article] [PubMed] [Google Scholar]289. Betancourt AM, Eltoum IA, Desmond RA, Russo J, Lamartiniere CA. 2010. In utero exposure to bisphenol A shifts the window of susceptibility for mammary carcinogenesis in the rat. Environ Health Perspect 118:1614–1619 [PMC free article] [PubMed] [Google Scholar]290. Weber Lozada K, Keri RA. 2011. Bisphenol A increases mammary cancer risk in two distinct mouse models of breast cancer. Biol Reprod 85:490–497 [PMC free article] [PubMed] [Google Scholar]291. Betancourt AM, Mobley JA, Russo J, Lamartiniere CA. 2010. Proteomic analysis in mammary glands of rat offspring exposed in utero to bisphenol A. J Proteomics 73:1241–1253 [PubMed] [Google Scholar]292. Lamartiniere CA, Jenkins S, Betancourt AM, Wang J, Russo J. 2011. Exposure to the endocrine disruptor bisphenol A alters susceptibility for mammary cancer. Horm Mol Biol Clin Investig 5:45–52 [PMC free article] [PubMed] [Google Scholar]293. Jenkins S, Wang J, Eltoum I, Desmond R, Lamartiniere CA. 2011. Chronic oral exposure to bisphenol A results in a non-monotonic dose response in mammary carcinogenesis and metastasis in MMTV-erbB2 mice. Environ Health Perspect 119:1604–1609 [PMC free article] [PubMed] [Google Scholar]294. Nikaido Y, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, Tsubura A. 2005. Effects of prepubertal exposure to xenoestrogen on development of estrogen target organs in female CD-1 mice. In Vivo 19:487–494 [PubMed] [Google Scholar]295. Yin H, Ito A, Bhattacharjee D, Hoshi M. 2006. A comparative study on the protective effects of 17β-estradiol, biochanin A and bisphenol A on mammary gland differentiation and tumorigenesis in rats. Indian J Exp Biol 44:540–546 [PubMed] [Google Scholar]296. Yang M, Ryu JH, Jeon R, Kang D, Yoo KY. 2009. Effects of bisphenol A on breast cancer and its risk factors. Arch Toxicol 83:281–285 [PubMed] [Google Scholar]297. Kortenkamp A. 2006. Breast cancer, oestrogens and environmental pollutants: a re-evaluation from a mixture perspective. Int J Androl 29:193–198 [PubMed] [Google Scholar]298. Hunt PA, Susiarjo M, Rubio C, Hassold TJ. 2009. The bisphenol A experience: a primer for the analysis of environmental effects on mammalian reproduction. Biol Reprod 81:807–813 [PMC free article] [PubMed] [Google Scholar]299. Carr R, Bertasi F, Betancourt A, Bowers S, Gandy BS, Ryan P, Willard S. 2003. Effect of neonatal rat bisphenol a exposure on performance in the Morris water maze. J Toxicol Environ Health A 66:2077–2088 [PubMed] [Google Scholar]300. Farabollini F, Porrini S, Dessì-Fulgherit F. 1999. Perinatal exposure to the estrogenic pollutant bisphenol A affects behavior in male and female rats. Pharmacol Biochem Behav 64:687–694 [PubMed] [Google Scholar]301. Fujimoto T, Kubo K, Aou S. 2006. Prenatal exposure to bisphenol A impairs sexual differentiation of exploratory behavior and increases depression-like behavior in rats. Brain Res 1068:49–55 [PubMed] [Google Scholar]302. Funabashi T, Kawaguchi M, Furuta M, Fukushima A, Kimura F. 2004. Exposure to bisphenol A during gestation and lactation causes loss of sex difference in corticotropin-releasing hormone-immunoreactive neurons in the bed nucleus of the stria terminalis of rats. Psychoneuroendocrinology 29:475–485 [PubMed] [Google Scholar]303. Kubo K, Arai O, Omura M, Watanabe R, Ogata R, Aou S. 2003. Low dose effects of bisphenol A on sexual differentiation of the brain and behavior in rats. Neurosci Res 45:345–356 [PubMed] [Google Scholar]304. Kubo K, Arai O, Ogata R, Omura M, Hori T, Aou S. 2001. Exposure to bisphenol A during the fetal and suckling periods disrupts sexual differentiation of the locus coeruleus and of behaviour in the rat. Neurosci Lett 304:73–76 [PubMed] [Google Scholar]305. Rubin BS, Lenkowski JR, Schaeberle CM, Vandenberg LN, Ronsheim PM, Soto AM. 2006. Evidence of altered brain sexual differentiation in mice exposed perinatally to low, environmentally relevant levels of bisphenol A. Endocrinology 147:3681–3691 [PubMed] [Google Scholar]306. Patisaul HB, Fortino AE, Polston EK. 2006. Neonatal genistein or bisphenol-A exposure alters sexual differentiation of the AVPV. Neurotoxicol Teratol 28:111–118 [PubMed] [Google Scholar]307. Adewale HB, Todd KL, Mickens JA, Patisaul HB. 2011. The impact of neonatal bisphenol: a exposure on sexually dimorphic hypothalamic nuclei in the female rat. Neurotoxicology 32:38–49 [PMC free article] [PubMed] [Google Scholar]308. Wolstenholme JT, Rissman EF, Connelly JJ. 2011. The role of bisphenol A in shaping the brain, epigenome and behavior. Horm Behav 59:296–305 [PMC free article] [PubMed] [Google Scholar]309. Maffini MV, Rubin BS, Sonnenschein C, Soto AM. 2006. Endocrine disruptors and reproductive health: the case of bisphenol-A. Mol Cell Endocrinol 254–255:179–186 [PubMed] [Google Scholar]310. Markey CM, Wadia PR, Rubin BS, Sonnenschein C, Soto AM. 2005. Long-term effects of fetal exposure to low doses of the xenoestrogen bisphenol-A in the female mouse genital tract. Biol Reprod 72:1344–1351 [PubMed] [Google Scholar]311. Yoshino S, Yamaki K, Li X, Sai T, Yanagisawa R, Takano H, Taneda S, Hayashi H, Mori Y. 2004. Prenatal exposure to bisphenol A up-regulates immune responses, including T helper 1 and T helper 2 responses, in mice. Immunology 112:489–495 [PMC free article] [PubMed] [Google Scholar]312. Yoshino S, Yamaki K, Yanagisawa R, Takano H, Hayashi H, Mori Y. 2003. Effects of bisphenol A on antigen-specific antibody production, proliferative responses of lymphoid cells, and TH1 and TH2 immune responses in mice. Br J Pharmacol 138:1271–1276 [PMC free article] [PubMed] [Google Scholar]313. Alonso-Magdalena P, Ropero AB, Soriano S, Quesada I, Nadal A. 2010. Bisphenol-A: a new diabetogenic factor? Hormones (Athens) 9:118–126 [PubMed] [Google Scholar]314. Rubin BS, Soto AM. 2009. Bisphenol A: perinatal exposure and body weight. Mol Cell Endocrinol 304:55–62 [PMC free article] [PubMed] [Google Scholar]315. Al-Hiyasat AS, Darmani H, Elbetieha AM. 2002. Effects of bisphenol A on adult male mouse fertility. Eur J Oral Sci 110:163–167 [PubMed] [Google Scholar]316. Cabaton NJ, Wadia PR, Rubin BS, Zalko D, Schaeberle CM, Askenase MH, Gadbois JL, Tharp AP, Whitt GS, Sonnenschein C, Soto AM. 2011. Perinatal exposure to environmentally relevant levels of bisphenol A decreases fertility and fecundity in CD-1 mice. Environ Health Perspect 119:547–552 [PMC free article] [PubMed] [Google Scholar]317. Al-Hiyasat AS, Darmani H, Elbetieha AM. 2004. Leached components from dental composites and their effects on fertility of female mice. Eur J Oral Sci 112:267–272 [PubMed] [Google Scholar]318. Salian S, Doshi T, Vanage G. 2009. Impairment in protein expression profile of testicular steroid receptor coregulators in male rat offspring perinatally exposed to Bisphenol A. Life Sci 85:11–18 [PubMed] [Google Scholar]319. Rubin BS. 2011. Bisphenol A: an endocrine disruptor with widespread exposure and multiple effects. J Steroid Biochem Mol Biol 127:27–34 [PubMed] [Google Scholar]320. Battaglin WA, Rice KC, Focazio MJ, Salmons S, Barry RX. 2009. The occurrence of glyphosate, atrazine, and other pesticides in vernal pools and adjacent streams in Washington, DC, Maryland, Iowa, and Wyoming, 2005–2006. Environ Monit Assess 155:281–307 [PubMed] [Google Scholar]321. Battaglin WA, Furlong ET, Burkhardt MR, Peter CJ. 2000. Occurrence of sulfonylurea, sulfonamide, imidazolinone, and other herbicides in rivers, reservoirs and ground water in the Midwestern United States, 1998. Sci Total Environ 248:123–133 [PubMed] [Google Scholar]322. Solomon KR, Baker DB, Richards RP, Dixon KR, Klaine SJ, La Point TW, Kendall RJ, Weisskopf CP, Giddings JM, Giesy JP, Hall LW, Jr, Williams M. 1996. Ecological risk assessment of atrazine in North American surface waters. Environ Toxicol Chem 15:31–76 [Google Scholar]323. Benachour N, Moslemi S, Sipahutar H, Seralini GE. 2007. Cytotoxic effects and aromatase inhibition by xenobiotic endocrine disrupters alone and in combination. Toxicol Appl Pharmacol 222:129–140 [PubMed] [Google Scholar]324. Sanderson JT, Seinen W, Giesy JP, van den Berg M. 2000. 2-Chloro-s-triazine herbicides induce aromatase (CYP19) activity in H295R human adrenocortical carcinoma cells: a novel mechanism for estrogenicity? Toxicol Sci 54:121–127 [PubMed] [Google Scholar]325. Sanderson JT, Letcher RJ, Heneweer M, Giesy JP, van den Berg M. 2001. Effects of chloro-s-triazine herbicides and metabolites on aromatase activity in various human cell lines and on vitellogenin production in male carp hepatocytes. Environ Health Perspect 109:1027–1031 [PMC free article] [PubMed] [Google Scholar]326. Hayes TB, Anderson LL, Beasley VR, de Solla SR, Iguchi T, Ingraham H, Kestemont P, Kniewald J, Kniewald Z, Langlois VS, Luque EH, McCoy KA, Muñoz-de-Toro M, Oka T, Oliveira CA, Orton F, Ruby S, Suzawa M, Tavera-Mendoza LE, Trudeau VL, Victor-Costa AB, Willingham E. 2011. Demasculinization and feminization of male gonads by atrazine: consistent effects across vertebrate classes. J Steroid Biochem Mol Biol 127:64–73 [PMC free article] [PubMed] [Google Scholar]327. Cooper RL, Laws SC, Das PC, Narotsky MG, Goldman JM, Lee Tyrey E, Stoker TE. 2007. Atrazine and reproductive function: mode and mechanism of action studies. Birth Defects Res B Dev Reprod Toxicol 80:98–112 [PubMed] [Google Scholar]328. Stoker TE, Robinette CL, Cooper RL. 1999. Maternal exposure to atrazine during lactation suppresses suckling-induced prolactin release and results in prostatitis in the adult offspring. Toxicol Sci 52:68–79 [PubMed] [Google Scholar]329. Laws SC, Hotchkiss M, Ferrell J, Jayaraman S, Mills L, Modic W, Tinfo N, Fraites M, Stoker T, Cooper R. 2009. Chlorotriazine herbicides and metabolites activate an ACTH-dependent release of corticosterone in male Wistar rats. Toxicol Sci 112:78–87 [PubMed] [Google Scholar]330. Fraites MJ, Cooper RL, Buckalew A, Jayaraman S, Mills L, Laws SC. 2009. Characterization of the hypothalamic-pituitary-adrenal axis response to atrazine and metabolites in the female rat. Toxicol Sci 112:88–99 [PubMed] [Google Scholar]331. Yoshimoto S, Okada E, Umemoto H, Tamura K, Uno Y, Nishida-Umehara C, Matsuda Y, Takamatsu N, Shiba T, Ito M. 2008. A W-linked DM-domain gene, DM-W, participates in primary ovary development in Xenopus laevis. Proc Natl Acad Sci USA 105:2469–2474 [PMC free article] [PubMed] [Google Scholar]332. Hayes TB. 1998. Sex determination and primary sex differentiation in amphibians. J Exp Zool 281:373–399 [PubMed] [Google Scholar]333. Ochoa-Acuña H, Frankenberger J, Hahn L, Carbajo C. 2009. Drinking-water herbicide exposure in Indiana and prevalence of small-for-gestational-age and preterm delivery. Environ Health Perspect 117:1619–1624 [PMC free article] [PubMed] [Google Scholar]334. Morgan MK, Scheuerman PR, Bishop CS, Pyles RA. 1996. Teratogenic potential of atrazine and 2,4-D using FETAX. J Toxicol Environ Health 48:151–168 [PubMed] [Google Scholar]335. Allran JW, Karasov WH. 2001. Effects of atrazine on embryos, larvae, and adults of anuran amphibians. Environ Toxicol Chem 20:769–775 [PubMed] [Google Scholar]336. Hayes TB, Collins A, Lee M, Mendoza M, Noriega N, Stuart AA, Vonk A. 2002. Hermaphroditic, demasculinized frogs after exposure to the herbicide atrazine at low ecologically relevant doses. Proc Natl Acad Sci USA 99:5476–5480 [PMC free article] [PubMed] [Google Scholar]337. Hayes TB, Khoury V, Narayan A, Nazir M, Park A, Brown T, Adame L, Chan E, Buchholz D, Stueve T, Gallipeau S. 2010. Atrazine induces complete feminization and chemical castration in male African clawed frogs (Xenopus laevis). Proc Natl Acad Sci USA 107:4612–4617 [PMC free article] [PubMed] [Google Scholar]338. Hayes TB, Stuart AA, Mendoza M, Collins A, Noriega N, Vonk A, Johnston G, Liu R, Kpodzo D. 2006. Characterization of atrazine-induced gonadal malformations in African clawed frogs (Xenopus laevis) and comparisons with effects of an androgen antagonist (cyproterone acetate) and exogenous estrogen (17β-estradiol): support for the demasculinization/feminization hypothesis. Environ Health Perspect 114:134–141 [PMC free article] [PubMed] [Google Scholar]339. Storrs-Méndez SI, Semlitsch RD. 2010. Intersex gonads in frogs: understanding the time course of natural development and role of endocrine disruptors. J Exp Zool B Mol Dev Evol 314:57–66 [PubMed] [Google Scholar]340. Carr JA, Gentles A, Smith EE, Goleman WL, Urquidi LJ, Thuett K, Kendall RJ, Giesy JP, Gross TS, Solomon KR, Van Der Kraak G. 2003. Response of larval Xenopus laevis to atrazine: assessment of growth, metamorphosis, and gonadal and laryngeal morphology. Environ Toxicol Chem 22:396–405 [PubMed] [Google Scholar]341. Hecker M, Kim WJ, Park JW, Murphy MB, Villeneuve D, Coady KK, Jones PD, Solomon KR, Van Der Kraak G, Carr JA, Smith EE, du Preez L, Kendall RJ, Giesy JP. 2005. Plasma concentrations of estradiol and testosterone, gonadal aromatase activity and ultrastructure of the testis in Xenopus laevis exposed to estradiol or atrazine. Aquat Toxicol 72:383–396 [PubMed] [Google Scholar]342. Orton F, Carr JA, Handy RD. 2006. Effects of nitrate and atrazine on larval development and sexual differentiation in the northern leopard frog Rana pipiens. Environ Toxicol Chem 25:65–71 [PubMed] [Google Scholar]343. Hayes T, Haston K, Tsui M, Hoang A, Haeffele C, Vonk A. 2003. Atrazine-induced hermaphroditism at 0.1 ppb in American leopard frogs (Rana pipiens): laboratory and field evidence. Environ Health Perspect 111:568–575 [PMC free article] [PubMed] [Google Scholar]344. Tavera-Mendoza L, Ruby S, Brousseau P, Fournier M, Cyr D, Marcogliese D. 2002. Response of the amphibian tadpole (Xenopus laevis) to atrazine during sexual differentiation of the testis. Environ Toxicol Chem 21:527–531 [PubMed] [Google Scholar]345. Oka T, Tooi O, Mitsui N, Miyahara M, Ohnishi Y, Takase M, Kashiwagi A, Shinkai T, Santo N, Iguchi T. 2008. Effect of atrazine on metamorphosis and sexual differentiation in Xenopus laevis. Aquat Toxicol 87:215–226 [PubMed] [Google Scholar]346. Langlois VS, Carew AC, Pauli BD, Wade MG, Cooke GM, Trudeau VL. 2010. Low levels of the herbicide atrazine alter sex ratios and reduce metamorphic success in Rana pipiens tadpoles raised in outdoor mesocosms. Environ Health Perspect 118:552–557 [PMC free article] [PubMed] [Google Scholar]347. Jooste AM, Du Preez LH, Carr JA, Giesy JP, Gross TS, Kendall RJ, Smith EE, Van der Kraak GL, Solomon KR. 2005. Gonadal development of larval male Xenopus laevis exposed to atrazine in outdoor microcosms. Environ Sci Technol 39:5255–5261 [PubMed] [Google Scholar]348. Spolyarich N, Hyne R, Wilson S, Palmer C, Byrne M. 2010. Growth, development and sex ratios of spotted marsh frog (Limnodynastes tasmaniensis) larvae exposed to atrazine and a herbicide mixture. Chemosphere 78:807–813 [PubMed] [Google Scholar]349. Hecker M, Park JW, Murphy MB, Jones PD, Solomon KR, Van Der Kraak G, Carr JA, Smith EE, du Preez L, Kendall RJ, Giesy JP. 2005. Effects of atrazine on CYP19 gene expression and aromatase activity in testes and on plasma sex steroid concentrations of male African clawed frogs (Xenopus laevis). Toxicol Sci 86:273–280 [PubMed] [Google Scholar]350. Du Preez LH, Kunene N, Everson GJ, Carr JA, Giesy JP, Gross TS, Hosmer AJ, Kendall RJ, Smith EE, Solomon KR, Van Der Kraak GJ. 2008. Reproduction, larval growth, and reproductive development in African clawed frogs (Xenopus laevis) exposed to atrazine. Chemosphere 71:546–552 [PubMed] [Google Scholar]351. Kloas W, Lutz I, Springer T, Krueger H, Wolf J, Holden L, Hosmer A. 2009. Does atrazine influence larval development and sexual differentiation in Xenopus laevis? Toxicol Sci 107:376–384 [PMC free article] [PubMed] [Google Scholar]352. U.S. Environmental Protection Agency 2010. October 9–12, 2007: The potential for atrazine to affect amphibian gonadal development. FIFRA Scientific Advisory Panel Meeting, Arlington, VA, 2007 [Google Scholar]353. McDaniel TV, Martin PA, Struger J, Sherry J, Marvin CH, McMaster ME, Clarence S, Tetreault G. 2008. Potential endocrine disruption of sexual development in free ranging male northern leopard frogs (Rana pipiens) and green frogs (Rana clamitans) from areas of intensive row crop agriculture. Aquat Toxicol 88:230–242 [PubMed] [Google Scholar]354. Reeder AL, Foley GL, Nichols DK, Hansen LG, Wikoff B, Faeh S, Eisold J, Wheeler MB, Warner R, Murphy JE, Beasley VR. 1998. Forms and prevalence of intersexuality and effects of environmental contaminants on sexuality in cricket frogs (Acris crepitans). Environ Health Perspect 106:261–266 [PMC free article] [PubMed] [Google Scholar]355. Hayes T, Haston K, Tsui M, Hoang A, Haeffele C, Vonk A. 2002. Feminization of male frogs in the wild. Nature 419:895–896 [PubMed] [Google Scholar]356. Spolyarich N, Hyne RV, Wilson SP, Palmer CG, Byrne M. 2011. Morphological abnormalities in frogs from a rice-growing region in NSW, Australia, with investigations into pesticide exposure. Environ Monit Assess 173:397–407 [PubMed] [Google Scholar]357. Du Preez LH, Kunene N, Hanner R, Giesy JP, Solomon KR, Hosmer A, Van Der Kraak GJ. 2009. Population-specific incidence of testicular ovarian follicles in Xenopus laevis from South Africa: a potential issue in endocrine testing. Aquat Toxicol 95:10–16 [PubMed] [Google Scholar]358. Murphy MB, Hecker M, Coady KK, Tompsett AR, Jones PD, Du Preez LH, Everson GJ, Solomon KR, Carr JA, Smith EE, Kendall RJ, Van Der Kraak G, Giesy JP. 2006. Atrazine concentrations, gonadal gross morphology and histology in ranid frogs collected in Michigan agricultural areas. Aquat Toxicol 76:230–245 [PubMed] [Google Scholar]359. Suzawa M, Ingraham HA. 2008. The herbicide atrazine activates endocrine gene networks via non-steroidal NR5A nuclear receptors in fish and mammalian cells. PLoS ONE 3:e2117. [PMC free article] [PubMed] [Google Scholar]360. Forson D, Storfer A. 2006. Effects of atrazine and iridovirus infection on survival and life-history traits of the long-toed salamander (Ambystoma macrodactylum). Environ Toxicol Chem 25:168–173 [PubMed] [Google Scholar]361. Forson DD, Storfer A. 2006. Atrazine increases ranavirus susceptibility in the tiger salamander, Ambystoma tigrinum. Ecol Appl 16:2325–2332 [PubMed] [Google Scholar]362. Rohr JR, Palmer BD. 2005. Aquatic herbicide exposure increases salamander desiccation risk eight months later in a terrestrial environment. Environ Toxicol Chem 24:1253–1258 [PubMed] [Google Scholar]363. Storrs SI, Kiesecker JM. 2004. Survivorship patterns of larval amphibians exposed to low concentrations of atrazine. Environ Health Perspect 112:1054–1057 [PMC free article] [PubMed] [Google Scholar]364. Nieves-Puigdoller K, Björnsson BT, McCormick SD. 2007. Effects of hexazinone and atrazine on the physiology and endocrinology of smolt development in Atlantic salmon. Aquat Toxicol 84:27–37 [PubMed] [Google Scholar]365. Barr DB, Panuwet P, Nguyen JV, Udunka S, Needham LL. 2007. Assessing exposure to atrazine and its metabolites using biomonitoring. Environ Health Perspect 115:1474–1478 [PMC free article] [PubMed] [Google Scholar]366. Curwin BD, Hein MJ, Sanderson WT, Striley C, Heederik D, Kromhout H, Reynolds SJ, Alavanja MC. 2007. Pesticide dose estimates for children of Iowa farmers and non-farmers. Environ Res 105:307–315 [PubMed] [Google Scholar]367. Rayner JL, Enoch RR, Fenton SE. 2005. Adverse effects of prenatal exposure to atrazine during a critical period of mammary gland growth. Toxicol Sci 87:255–266 [PubMed] [Google Scholar]368. Rayner JL, Wood C, Fenton SE. 2004. Exposure parameters necessary for delayed puberty and mammary gland development in Long-Evans rats exposed in utero to atrazine. Toxicol Appl Pharmacol 195:23–34 [PubMed] [Google Scholar]369. Cooper RL, Stoker TE, Goldman JM, Parrish MB, Tyrey L. 1996. Effect of atrazine on ovarian function in the rat. Reprod Toxicol 10:257–264 [PubMed] [Google Scholar]370. Friedmann AS. 2002. Atrazine inhibition of testosterone production in rat males following peripubertal exposure. Reprod Toxicol 16:275–279 [PubMed] [Google Scholar]371. Rayner JL, Enoch RR, Wolf DC, Fenton SE. 2007. Atrazine-induced reproductive tract alterations after transplacental and/or lactational exposure in male Long-Evans rats. Toxicol Appl Pharmacol 218:238–248 [PubMed] [Google Scholar]372. Karrow NA, McCay JA, Brown RD, Musgrove DL, Guo TL, Germolec DR, White KL., Jr 2005. Oral exposure to atrazine modulates cell-mediated immune function and decreases host resistance to the B16F10 tumor model in female B6C3F1 mice. Toxicology 209:15–28 [PubMed] [Google Scholar]373. Enoch RR, Stanko JP, Greiner SN, Youngblood GL, Rayner JL, Fenton SE. 2007. Mammary gland development as a sensitive end point after acute prenatal exposure to an atrazine metabolite mixture in female Long-Evans rats. Environ Health Perspect 115:541–547 [PMC free article] [PubMed] [Google Scholar]374. Stanko JP, Enoch RR, Rayner JL, Davis CC, Wolf DC, Malarkey DE, Fenton SE. 2010. Effects of prenatal exposure to a low dose atrazine metabolite mixture on pubertal timing and prostate development of male Long-Evans rats. Reprod Toxicol 30:540–549 [PMC free article] [PubMed] [Google Scholar]375. Schecter A, Birnbaum L, Ryan JJ, Constable JD. 2006. Dioxins: an overview. Environ Res 101:419–428 [PubMed] [Google Scholar]376. Mukerjee D. 1998. Health impact of polychlorinated dibenzo-p-dioxins: a critical review. J Air Waste Manag Assoc 48:157–165 [PubMed] [Google Scholar]377. Emond C, Birnbaum LS, DeVito MJ. 2006. Use of a physiologically based pharmacokinetic model for rats to study the influence of body fat mass and induction of CYP1A2 on the pharmacokinetics of TCDD. Environ Health Perspect 114:1394–1400 [PMC free article] [PubMed] [Google Scholar]378. Milbrath MO, Wenger Y, Chang CW, Emond C, Garabrant D, Gillespie BW, Jolliet O. 2009. Apparent half-lives of dioxins, furans, and polychlorinated biphenyls as a function of age, body fat, smoking status, and breast-feeding. Environ Health Perspect 117:417–425 [PMC free article] [PubMed] [Google Scholar]379. Emond C, Michalek JE, Birnbaum LS, DeVito MJ. 2005. Comparison of the use of a physiologically based pharmacokinetic model and a classical pharmacokinetic model for dioxin exposure assessments. Environ Health Perspect 113:1666–1668 [PMC free article] [PubMed] [Google Scholar]380. Gierthy JF, Crane D. 1984. Reversible inhibition of in vitro epithelial cell proliferation by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol 74:91–98 [PubMed] [Google Scholar]381. Korkalainen M, Tuomisto J, Pohjanvirta R. 2001. The AH receptor of the most dioxin-sensitive species, guinea pig, is highly homologous to the human AH receptor. Biochem Biophys Res Commun 285:1121–1129 [PubMed] [Google Scholar]382. Okey AB, Riddick DS, Harper PA. 1994. The Ah receptor: mediator of the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and related compounds. Toxicol Lett 70:1–22 [PubMed] [Google Scholar]383. Matsumura F. 2009. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem Pharmacol 77:608–626 [PubMed] [Google Scholar]384. Birnbaum LS, Tuomisto J. 2000. Non-carcinogenic effects of TCDD in animals. Food Addit Contam 17:275–288 [PubMed] [Google Scholar]385. DeVito MJ, Birnbaum LS, Farland WH, Gasiewicz TA. 1995. Comparisons of estimated human body burdens of dioxinlike chemicals and TCDD body burdens in experimentally exposed animals. Environ Health Perspect 103:820–831 [PMC free article] [PubMed] [Google Scholar]386. Kung T, Murphy KA, White LA. 2009. The aryl hydrocarbon receptor (AhR) pathway as a regulatory pathway for cell adhesion and matrix metabolism. Biochem Pharmacol 77:536–546 [PMC free article] [PubMed] [Google Scholar]387. Li H, Wang H. 2010. Activation of xenobiotic receptors: driving into the nucleus. Expert Opin Drug Metab Toxicol 6:409–426 [PMC free article] [PubMed] [Google Scholar]388. Marinkoviæ N, Pašaliæ D, Ferenèak G, Grškoviæ B, Stavljeniæ Rukavina A. 2010. Dioxins and human toxicity. Arh Hig Rada Toksikol 61:445–453 [PubMed] [Google Scholar]389. White SS, Birnbaum LS. 2009. An overview of the effects of dioxins and dioxin-like compounds on vertebrates, as documented in human and ecological epidemiology. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 27:197–211 [PMC free article] [PubMed] [Google Scholar]390. Swedenborg E, Pongratz I. 2010. AhR and ARNT modulate ER signaling. Toxicology 268:132–138 [PubMed] [Google Scholar]391. Schwetz BA, Norris JM, Sparschu GL, Rowe UK, Gehring PJ, Emerson JL, Gerbig CG. 1973. Toxicology of chlorinated dibenzo-p-dioxins. Environ Health Perspect 5:87–99 [PMC free article] [PubMed] [Google Scholar]392. Kociba RJ, Schwetz BA. 1982. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Drug Metab Rev 13:387–406 [PubMed] [Google Scholar]393. Couture LA, Abbott BD, Birnbaum LS. 1990. A critical review of the developmental toxicity and teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin: recent advances toward understanding the mechanism. Teratology 42:619–627 [PubMed] [Google Scholar]394. Mocarelli P, Needham LL, Marocchi A, Patterson DG, Jr, Brambilla P, Gerthoux PM, Meazza L, Carreri V. 1991. Serum concentrations of 2,3,7,8-tetrachlorodibenzo-p-dioxin and test results from selected residents of Seveso, Italy. J Toxicol Environ Health 32:357–366 [PubMed] [Google Scholar]395. Geusau A, Abraham K, Geissler K, Sator MO, Stingl G, Tschachler E. 2001. Severe 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) intoxication: clinical and laboratory effects. Environ Health Perspect 109:865–869 [PMC free article] [PubMed] [Google Scholar]396. Pohjanvirta R, Tuomisto J. 1994. Short-term toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in laboratory animals: effects, mechanisms, and animal models. Pharmacol Rev 46:483–549 [PubMed] [Google Scholar]397. Chahoud I, Hartmann J, Rune GM, Neubert D. 1992. Reproductive toxicity and toxicokinetics of 2,3,7,8-tetrachlorodibenzo-p-dioxin. 3. Effects of single doses on the testis of male rats. Arch Toxicol 66:567–572 [PubMed] [Google Scholar]398. Mocarelli P, Gerthoux PM, Needham LL, Patterson DG, Jr, Limonta G, Falbo R, Signorini S, Bertona M, Crespi C, Sarto C, Scott PK, Turner WE, Brambilla P. 2011. Perinatal exposure to low doses of dioxin can permanently impair human semen quality. Environ Health Perspect 119:713–718 [PMC free article] [PubMed] [Google Scholar]399. Mocarelli P, Gerthoux PM, Patterson DG, Jr, Milani S, Limonta G, Bertona M, Signorini S, Tramacere P, Colombo L, Crespi C, Brambilla P, Sarto C, Carreri V, Sampson EJ, Turner WE, Needham LL. 2008. Dioxin exposure, from infancy through puberty, produces endocrine disruption and affects human semen quality. Environ Health Perspect 116:70–77 [PMC free article] [PubMed] [Google Scholar]400. Foster WG, Maharaj-Briceño S, Cyr DG. 2010. Dioxin-induced changes in epididymal sperm count and spermatogenesis. Environ Health Perspect 118:458–464 [PMC free article] [PubMed] [Google Scholar]401. Bell DR, Clode S, Fan MQ, Fernandes A, Foster PM, Jiang T, Loizou G, MacNicoll A, Miller BG, Rose M, Tran L, White S. 2010. Interpretation of studies on the developmental reproductive toxicology of 2,3,7,8-tetrachlorodibenzo-p-dioxin in male offspring. Food Chem Toxicol 48:1439–1447 [PMC free article] [PubMed] [Google Scholar]402. Bjerke DL, Peterson RE. 1994. Reproductive toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in male rats: different effects of in utero versus lactational exposure. Toxicol Appl Pharmacol 127:241–249 [PubMed] [Google Scholar]403. Faqi AS, Dalsenter PR, Merker HJ, Chahoud I. 1998. Reproductive toxicity and tissue concentrations of low doses of 2,3,7,8-tetrachlorodibenzo-p-dioxin in male offspring rats exposed throughout pregnancy and lactation. Toxicol Appl Pharmacol 150:383–392 [PubMed] [Google Scholar]404. Gray LE, Jr, Kelce WR, Monosson E, Ostby JS, Birnbaum LS. 1995. Exposure to TCDD during development permanently alters reproductive function in male Long Evans rats and hamsters: reduced ejaculated and epididymal sperm numbers and sex accessory gland weights in offspring with normal androgenic status. Toxicol Appl Pharmacol 131:108–118 [PubMed] [Google Scholar]405. Gray LE, Ostby JS, Kelce WR. 1997. A dose-response analysis of the reproductive effects of a single gestational dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin in male Long Evans hooded rat offspring. Toxicol Appl Pharmacol 146:11–20 [PubMed] [Google Scholar]406. Ohsako S, Miyabara Y, Sakaue M, Ishimura R, Kakeyama M, Izumi H, Yonemoto J, Tohyama C. 2002. Developmental stage-specific effects of perinatal 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on reproductive organs of male rat offspring. Toxicol Sci 66:283–292 [PubMed] [Google Scholar]407. Simanainen U, Haavisto T, Tuomisto JT, Paranko J, Toppari J, Tuomisto J, Peterson RE, Viluksela M. 2004. Pattern of male reproductive system effects after in utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure in three differentially TCDD-sensitive rat lines. Toxicol Sci 80:101–108 [PubMed] [Google Scholar]408. Sommer RJ, Ippolito DL, Peterson RE. 1996. In utero and lactational exposure of the male Holtzman rat to 2,3,7,8-tetrachlorodibenzo-p-dioxin: decreased epididymal and ejaculated sperm numbers without alterations in sperm transit rate. Toxicol Appl Pharmacol 140:146–153 [PubMed] [Google Scholar]409. Mably TA, Bjerke DL, Moore RW, Gendron-Fitzpatrick A, Peterson RE. 1992. In utero and lactational exposure of male rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin. 3. Effects on spermatogenesis and reproductive capability. Toxicol Appl Pharmacol 114:118–126 [PubMed] [Google Scholar]410. Wilker C, Johnson L, Safe S. 1996. Effects of developmental exposure to indole-3-carbinol or 2,3,7,8-tetrachlorodibenzo-p-dioxin on reproductive potential of male rat offspring. Toxicol Appl Pharmacol 141:68–75 [PubMed] [Google Scholar]411. Jin MH, Hong CH, Lee HY, Kang HJ, Han SW. 2010. Toxic effects of lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on development of male reproductive system: involvement of antioxidants, oxidants, and p53 protein. Environ Toxicol 25:1–8 [PubMed] [Google Scholar]412. Loeffler IK, Peterson RE. 1999. Interactive effects of TCDD and p,p′-DDE on male reproductive tract development in in utero and lactationally exposed rats. Toxicol Appl Pharmacol 154:28–39 [PubMed] [Google Scholar]413. Rebourcet D, Odet F, Vérot A, Combe E, Meugnier E, Pesenti S, Leduque P, Déchaud H, Magre S, Le Magueresse-Battistoni B. 2010. The effects of an in utero exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on male reproductive function: identification of Ccl5 as a potential marker. Int J Androl 33:413–424 [PMC free article] [PubMed] [Google Scholar]414. Bell DR, Clode S, Fan MQ, Fernandes A, Foster PM, Jiang T, Loizou G, MacNicoll A, Miller BG, Rose M, Tran L, White S. 2007. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the developing male Wistar(Han) rat. I. No decrease in epididymal sperm count after a single acute dose. Toxicol Sci 99:214–223 [PubMed] [Google Scholar]415. Bell DR, Clode S, Fan MQ, Fernandes A, Foster PM, Jiang T, Loizou G, MacNicoll A, Miller BG, Rose M, Tran L, White S. 2007. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the developing male Wistar(Han) rat. II. Chronic dosing causes developmental delay. Toxicol Sci 99:224–233 [PubMed] [Google Scholar]416. Ohsako S, Miyabara Y, Nishimura N, Kurosawa S, Sakaue M, Ishimura R, Sato M, Takeda K, Aoki Y, Sone H, Tohyama C, Yonemoto J. 2001. Maternal exposure to a low dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) suppressed the development of reproductive organs of male rats: dose-dependent increase of mRNA levels of 5α-reductase type 2 in contrast to decrease of androgen receptor in the pubertal ventral prostate. Toxicol Sci 60:132–143 [PubMed] [Google Scholar]417. Yonemoto J, Ichiki T, Takei T, Tohyama C. 2005. Maternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin and the body burden in offspring of Long-Evans rats. Environ Health Prev Med 10:21–32 [PMC free article] [PubMed] [Google Scholar]418. Arima A, Kato H, Ooshima Y, Tateishi T, Inoue A, Muneoka A, Ihara T, Kamimura S, Fukusato T, Kubota S, Sumida H, Yasuda M. 2009. In utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces a reduction in epididymal and ejaculated sperm number in rhesus monkeys. Reprod Toxicol 28:495–502 [PubMed] [Google Scholar]419. Yamano Y, Asano A, Ohta M, Hirata S, Shoda T, Ohyama K. 2009. Expression of rat sperm flagellum-movement associated protein genes under 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment. Biosci Biotechnol Biochem 73:946–949 [PubMed] [Google Scholar]420. Korkalainen M, Tuomisto J, Pohjanvirta R. 2004. Primary structure and inducibility by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) of aryl hydrocarbon receptor repressor in a TCDD-sensitive and a TCDD-resistant rat strain. Biochem Biophys Res Commun 315:123–131 [PubMed] [Google Scholar]421. Ishimaru N, Takagi A, Kohashi M, Yamada A, Arakaki R, Kanno J, Hayashi Y. 2009. Neonatal exposure to low-dose 2,3,7,8-tetrachlorodibenzo-p-dioxin causes autoimmunity due to the disruption of T cell tolerance. J Immunol 182:6576–6586 [PubMed] [Google Scholar]422. Nohara K, Fujimaki H, Tsukumo S, Ushio H, Miyabara Y, Kijima M, Tohyama C, Yonemoto J. 2000. The effects of perinatal exposure to low doses of 2,3,7,8-tetrachlorodibenzo-p-dioxin on immune organs in rats. Toxicology 154:123–133 [PubMed] [Google Scholar]423. Lim J, DeWitt JC, Sanders RA, Watkins JB, 3rd, Henshel DS. 2007. Suppression of endogenous antioxidant enzymes by 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced oxidative stress in chicken liver during development. Arch Environ Contam Toxicol 52:590–595 [PubMed] [Google Scholar]424. Slezak BP, Hatch GE, DeVito MJ, Diliberto JJ, Slade R, Crissman K, Hassoun E, Birnbaum LS. 2000. Oxidative stress in female B6C3F1 mice following acute and subchronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol Sci 54:390–398 [PubMed] [Google Scholar]425. Hassoun EA, Wilt SC, Devito MJ, Van Birgelen A, Alsharif NZ, Birnbaum LS, Stohs SJ. 1998. Induction of oxidative stress in brain tissues of mice after subchronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci 42:23–27 [PubMed] [Google Scholar]426. Hermsen SA, Larsson S, Arima A, Muneoka A, Ihara T, Sumida H, Fukusato T, Kubota S, Yasuda M, Lind PM. 2008. In utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) affects bone tissue in rhesus monkeys. Toxicology 253:147–152 [PubMed] [Google Scholar]427. Keller JM, Huet-Hudson Y, Leamy LJ. 2008. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on molar development among non-resistant inbred strains of mice: a geometric morphometric analysis. Growth Dev Aging 71:3–16 [PubMed] [Google Scholar]428. Kakeyama M, Sone H, Tohyama C. 2008. Perinatal exposure of female rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin induces central precocious puberty in the offspring. J Endocrinol 197:351–358 [PubMed] [Google Scholar]429. Shi Z, Valdez KE, Ting AY, Franczak A, Gum SL, Petroff BK. 2007. Ovarian endocrine disruption underlies premature reproductive senescence following environmentally relevant chronic exposure to the aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biol Reprod 76:198–202 [PubMed] [Google Scholar]430. Gray LE, Wolf C, Mann P, Ostby JS. 1997. In utero exposure to low doses of 2,3,7,8-tetrachlorodibenzo-p-dioxin alters reproductive development of female Long Evans hooded rat offspring. Toxicol Appl Pharmacol 146:237–244 [PubMed] [Google Scholar]431. Jenkins S, Rowell C, Wang J, Lamartiniere CA. 2007. Prenatal TCDD exposure predisposes for mammary cancer in rats. Reprod Toxicol 23:391–396 [PMC free article] [PubMed] [Google Scholar]432. Mitsui T, Sugiyama N, Maeda S, Tohyama C, Arita J. 2006. Perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin suppresses contextual fear conditioning-accompanied activation of cyclic AMP response element-binding protein in the hippocampal CA1 region of male rats. Neurosci Lett 398:206–210 [PubMed] [Google Scholar]433. Seo BW, Powers BE, Widholm JJ, Schantz SL. 2000. Radial arm maze performance in rats following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Neurotoxicol Teratol 22:511–519 [PubMed] [Google Scholar]434. Uemura H, Arisawa K, Hiyoshi M, Kitayama A, Takami H, Sawachika F, Dakeshita S, Nii K, Satoh H, Sumiyoshi Y, Morinaga K, Kodama K, Suzuki T, Nagai M, Suzuki T. 2009. Prevalence of metabolic syndrome associated with body burden levels of dioxin and related compounds among Japan’s general population. Environ Health Perspect 117:568–573 [PMC free article] [PubMed] [Google Scholar]435. Hites RA. 2011. Dioxins: an overview and history. Environ Sci Technol 45:16–20 [PubMed] [Google Scholar]436. De Groef B, Decallonne BR, Van der Geyten S, Darras VM, Bouillon R. 2006. Perchlorate versus other environmental sodium/iodide symporter inhibitors: potential thyroid-related health effects. Eur J Endocrinol 155:17–25 [PubMed] [Google Scholar]437. Blount BC, Valentin-Blasini L, Osterloh JD, Mauldin JP, Pirkle JL. 2007. Perchlorate exposure of the US Population, 2001–2002. J Expo Sci Environ Epidemiol 17:400–407 [PubMed] [Google Scholar]438. Greer MA, Goodman G, Pleus RC, Greer SE. 2002. Health effects assessment for environmental perchlorate contamination: the dose response for inhibition of thyroidal radioiodine uptake in humans. Environ Health Perspect 110:927–937 [PMC free article] [PubMed] [Google Scholar]439. Murray CW, Egan SK, Kim H, Beru N, Bolger PM. 2008. US Food and Drug Administration’s Total Diet Study: Dietary intake of perchlorate and iodine. J Expo Sci Environ Epidemiol 18:571–580 [PubMed] [Google Scholar]440. Huber DR, Blount BC, Mage DT, Letkiewicz FJ, Kumar A, Allen RH. 2011. Estimating perchlorate exposure from food and tap water based on US biomonitoring and occurrence data. J Expo Sci Environ Epidemiol 21:395–407 [PubMed] [Google Scholar]441. Urbansky ET. 2002. Perchlorate as an environmental contaminant. Environ Sci Pollut Res Int 9:187–192 [PubMed] [Google Scholar]442. Ginsberg GL, Hattis DB, Zoeller RT, Rice DC. 2007. Evaluation of the U.S. EPA/OSWER preliminary remediation goal for perchlorate in groundwater: focus on exposure to nursing infants. Environ Health Perspect 115:361–369 [PMC free article] [PubMed] [Google Scholar]443. Dasgupta PK, Dyke JV, Kirk AB, Jackson WA. 2006. Perchlorate in the United States. Analysis of relative source contributions to the food chain. Environ Sci Technol 40:6608–6614 [PubMed] [Google Scholar]444. Tan K, Anderson TA, Jones MW, Smith PN, Jackson WA. 2004. Accumulation of perchlorate in aquatic and terrestrial plants at a field scale. J Environ Qual 33:1638–1646 [PubMed] [Google Scholar]445. Miller MD, Crofton KM, Rice DC, Zoeller RT. 2009. Thyroid-disrupting chemicals: interpreting upstream biomarkers of adverse outcomes. Environ Health Perspect 117:1033–1041 [PMC free article] [PubMed] [Google Scholar]446. Wolff J. 1998. Perchlorate and the thyroid gland. Pharmacol Rev 50:89–105 [PubMed] [Google Scholar]447. Carrasco N. 2000. Thyroid iodide transport: the Na+/I− symporter (NIS). In: Braverman LE, Utiger RD, eds. The thyroid: a fundamental and clinical text. 8th ed Philidelphia: Lippincott, Williams and Wilkins; 52–61 [Google Scholar]448. Nicola JP, Basquin C, Portulano C, Reyna-Neyra A, Paroder M, Carrasco N. 2009. The Na+/I− symporter mediates active iodide uptake in the intestine. Am J Physiol Cell Physiol 296:C654–C662 [PMC free article] [PubMed] [Google Scholar]449. Vayre L, Sabourin JC, Caillou B, Ducreux M, Schlumberger M, Bidart JM. 1999. Immunohistochemical analysis of Na+/I− symporter distribution in human extra-thyroidal tissues. Eur J Endocrinol 141:382–386 [PubMed] [Google Scholar]450. 2007. The Na+/I symporter (NIS) mediates electroneutral active transport of the environmental pollutant perchlorate. Proc Natl Acad Sci USA 104:20250–20255 [PMC free article] [PubMed] [Google Scholar]451. Dohan O, De la Vieja A, Paroder V, Riedel C, Artani M, Reed M, Ginter CS, Carrasco N. 2003. The sodium/iodide symporter (NIS): characterization, regulation, and medical significance. Endocr Rev 24:48–77 [PubMed] [Google Scholar]452. Mitchell AM, Manley SW, Morris JC, Powell KA, Bergert ER, Mortimer RH. 2001. Sodium iodide symporter (NIS) gene expression in human placenta. Placenta 22:256–258 [PubMed] [Google Scholar]453. Szinnai G, Lacroix L, Carré A, Guimiot F, Talbot M, Martinovic J, Delezoide AL, Vekemans M, Michiels S, Caillou B, Schlumberger M, Bidart JM, Polak M. 2007. Sodium/iodide symporter (NIS) gene expression is the limiting step for the onset of thyroid function in the human fetus. J Clin Endocrinol Metab 92:70–76 [PubMed] [Google Scholar]454. Blount BC, Rich DQ, Valentin-Blasini L, Lashley S, Ananth CV, Murphy E, Smulian JC, Spain BJ, Barr DB, Ledoux T, Hore P, Robson M. 2009. Perinatal exposure to perchlorate. thiocyanate, and nitrate in New Jersey mothers and newborns. Environ Sci Technol 43:7543–7549 [PMC free article] [PubMed] [Google Scholar]455. Blount BC, Valentin-Blasini L. 2006. Analysis of perchlorate, thiocyanate, nitrate and iodide in human amniotic fluid using ion chromatography and electrospray tandem mass spectrometry. Anal Chim Acta 567:87–93 [PubMed] [Google Scholar]456. Borjan M, Marcella S, Blount B, Greenberg M, Zhang JJ, Murphy E, Valentin-Blasini L, Robson M. 2011. Perchlorate exposure in lactating women in an urban community in New Jersey. Sci Total Environ 409:460–464 [PubMed] [Google Scholar]457. Schier JG, Wolkin AF, Valentin-Blasini L, Belson MG, Kieszak SM, Rubin CS, Blount BC. 2010. Perchlorate exposure from infant formula and comparisons with the perchlorate reference dose. J Expo Sci Environ Epidemiol 20:281–287 [PubMed] [Google Scholar]458. Pearce EN, Leung AM, Blount BC, Bazrafshan HR, He X, Pino S, Valentin-Blasini L, Braverman LE. 2007. Breast milk iodine and perchlorate concentrations in lactating Boston-area women. J Clin Endocrinol Metab 92:1673–1677 [PubMed] [Google Scholar]459. Kirk AB, Dyke JV, Martin CF, Dasgupta PK. 2007. Temporal patterns in perchlorate, thiocyanate, and iodide excretion in human milk. Environ Health Perspect 115:182–186 [PMC free article] [PubMed] [Google Scholar]460. Zoeller RT, Rovet J. 2004. Timing of thyroid hormone action in the developing brain: clinical observations and experimental findings. J Neuroendocrinol 16:809–818 [PubMed] [Google Scholar]461. Ghassabian A, Bongers-Schokking JJ, Henrichs J, Jaddoe VW, Visser TJ, Visser W, de Muinck Keizer-Schrama SM, Hooijkaas H, Steegers EA, Hofman A, Verhulst FC, van der Ende J, de Rijke YB, Tiemeier H. 2011. Maternal thyroid function during pregnancy and behavioral problems in the offspring: the generation R study. Pediatr Res 69:454–459 [PubMed] [Google Scholar]462. Ghassabian A, Bongers-Schokking JJ, Henrichs J, Jaddoe VW, Visser TJ, Visser W, de Muinck Keizer-Schrama SM, Hooijkaas H, Steegers EA, Hofman A, Verhulst FC, van den Ende J, de Rijke YB, Tiemeier H. 2011. Maternal thyroid function during pregnancy and parent-report problem behavior of the offspring up to age three years. The Generation R Study. Pediatr Res 69(5 Pt 1):454–459 [PubMed] [Google Scholar]463. Murcia M, Rebagliato M, Iñiguez C, Lopez-Espinosa MJ, Estarlich M, Plaza B, Barona-Vilar C, Espada M, Vioque J, Ballester F. 2011. Effect of iodine supplementation during pregnancy on infant neurodevelopment at 1 year of age. Am J Epidemiol 173:804–812 [PubMed] [Google Scholar]464. Lawrence J, Lamm S, Braverman LE. 2001. Low dose perchlorate (3 mg daily) and thyroid function. Thyroid 11:295. [PubMed] [Google Scholar]465. Lawrence JE, Lamm SH, Pino S, Richman K, Braverman LE. 2000. The effect of short-term low-dose perchlorate on various aspects of thyroid function. Thyroid 10:659–663 [PubMed] [Google Scholar]466. Braverman LE, Pearce EN, He X, Pino S, Seeley M, Beck B, Magnani B, Blount BC, Firek A. 2006. Effects of six months of daily low-dose perchlorate exposure on thyroid function in healthy volunteers. J Clin Endocrinol Metab 91:2721–2724 [PubMed] [Google Scholar]467. National Research Council 2005. Health implications of perchlorate ingestion. Washington, DC: National Academies Press [Google Scholar]468. Eskenazi B, Warner M, Marks AR, Samuels S, Gerthoux PM, Vercellini P, Olive DL, Needham L, Patterson D, Jr, Mocarelli P. 2005. Serum dioxin concentrations and age at menopause. Environ Health Perspect 113:858–862 [PMC free article] [PubMed] [Google Scholar]469. Bleys J, Navas-Acien A, Laclaustra M, Pastor-Barriuso R, Menke A, Ordovas J, Stranges S, Guallar E. 2009. Serum selenium and peripheral arterial disease: results from the national health and nutrition examination survey, 2003–2004. Am J Epidemiol 169:996–1003 [PMC free article] [PubMed] [Google Scholar]470. Hatch EE, Nelson JW, Qureshi MM, Weinberg J, Moore LL, Singer M, Webster TF. 2008. Body mass index and waist circumference: a cross-sectional study of NHANES data, 1999–2002. Environ Health 7:27. [PMC free article] [PubMed] [Google Scholar]471. Brucker-Davis F, Thayer K, Colborn T, Fenichel P. 2002. Perchlorate: low dose exposure and susceptible populations. Thyroid 12:739; author reply 739–740 [PubMed] [Google Scholar]472. Gibbs JP, Ahmad R, Crump KS, Houck DP, Leveille TS, Findley JE, Francis M. 1998. Evaluation of a population with occupational exposure to airborne ammonium perchlorate for possible acute or chronic effects on thyroid function. J Occup Environ Med 40:1072–1082 [PubMed] [Google Scholar]473. Lamm SH, Braverman LE, Li FX, Richman K, Pino S, Howearth G. 1999. Thyroid health status of ammonium perchlorate workers: a cross-sectional occupational health study. J Occup Environ Med 41:248–260 [PubMed] [Google Scholar]474. Braverman LE, He X, Pino S, Cross M, Magnani B, Lamm SH, Kruse MB, Engel A, Crump KS, Gibbs JP. 2005. The effect of perchlorate, thiocyanate, and nitrate on thyroid function in workers exposed to perchlorate long-term. J Clin Endocrinol Metab 90:700–706 [PubMed] [Google Scholar]475. Blount BC, Pirkle JL, Osterloh JD, Valentin-Blasini L, Caldwell KL. 2006. Urinary perchlorate and thyroid hormone levels in adolescent and adult men and women living in the United States. Environ Health Perspect 114:1865–1871 [PMC free article] [PubMed] [Google Scholar]476. LaFranchi SH, Austin J. 2007. How should we be treating children with congenital hypothyroidism? J Pediatr Endocrinol Metab 20:559–578 [PubMed] [Google Scholar]477. Steinmaus C, Miller MD, Howd R. 2007. Impact of smoking and thiocyanate on perchlorate and thyroid hormone associations in the 2001–2002 national health and nutrition examination survey. Environ Health Perspect 115:1333–1338 [PMC free article] [PubMed] [Google Scholar]478. Li Z, Li FX, Byrd D, Deyhle GM, Sesser DE, Skeels MR, Lamm SH. 2000. Neonatal thyroxine level and perchlorate in drinking water. J Occup Environ Med 42:200–205 [PubMed] [Google Scholar]479. Li FX, Byrd DM, Deyhle GM, Sesser DE, Skeels MR, Katkowsky SR, Lamm SH. 2000. Neonatal thyroid-stimulating hormone level and perchlorate in drinking water. Teratology 62:429–431 [PubMed] [Google Scholar]480. Lamm SH, Doemland M. 1999. Has perchlorate in drinking water increased the rate of congenital hypothyroidism? J Occup Environ Med 41:409–411 [PubMed] [Google Scholar]481. Téllez Téllez R, Michaud Chacón P, Reyes Abarca C, Blount BC, Van Landingham CB, Crump KS, Gibbs JP. 2005. Long-term environmental exposure to perchlorate through drinking water and thyroid function during pregnancy and the neonatal period. Thyroid 15:963–975 [PubMed] [Google Scholar]482. Buffler PA, Kelsh MA, Lau EC, Edinboro CH, Barnard JC, Rutherford GW, Daaboul JJ, Palmer L, Lorey FW. 2006. Thyroid function and perchlorate in drinking water: an evaluation among California newborns, 1998. Environ Health Perspect 114:798–804 [PMC free article] [PubMed] [Google Scholar]483. Kelsh MA, Buffler PA, Daaboul JJ, Rutherford GW, Lau EC, Barnard JC, Exuzides AK, Madl AK, Palmer LG, Lorey FW. 2003. Primary congenital hypothyroidism, newborn thyroid function, and environmental perchlorate exposure among residents of a southern California community. J Occup Environ Med 45:1116–1127 [PubMed] [Google Scholar]484. Amitai Y, Winston G, Sack J, Wasser J, Lewis M, Blount BC, Valentin-Blasini L, Fisher N, Israeli A, Leventhal A. 2007. Gestational exposure to high perchlorate concentrations in drinking water and neonatal thyroxine levels. Thyroid 17:843–850 [PubMed] [Google Scholar]485. Steinmaus C, Miller MD, Smith AH. 2010. Perchlorate in drinking water during pregnancy and neonatal thyroid hormone levels in California. J Occup Environ Med 52:1217–1524 [PubMed] [Google Scholar]486. Brechner RJ, Parkhurst GD, Humble WO, Brown MB, Herman WH. 2000. Ammonium perchlorate contamination of Colorado River drinking water is associated with abnormal thyroid function in newborns in Arizona. J Occup Environ Med 42:777–782 [PubMed] [Google Scholar]487. Crump C, Michaud P, Téllez R, Reyes C, Gonzalez G, Montgomery EL, Crump KS, Lobo G, Becerra C, Gibbs JP. 2000. Does perchlorate in drinking water affect thyroid function in newborns or school-age children? J Occup Environ Med 42:603–612 [PubMed] [Google Scholar]488. Pearce EN, Spencer CA, Mestman JH, Lee RH, Bergoglio LM, Mereshian P, He X, Leung AM, Braverman LE. 2011. The effect of environmental perchlorate on thyroid function in pregnant women from Cordoba, Argentina, and Los Angeles, California. Endocr Pract 17:412–417 [PubMed] [Google Scholar]489. Pearce EN, Lazarus JH, Smyth PP, He X, Dall’amico D, Parkes AB, Burns R, Smith DF, Maina A, Bestwick JP, Jooman M, Leung AM, Braverman LE. 2010. Perchlorate and thiocyanate exposure and thyroid function in first-trimester pregnant women. J Clin Endocrinol Metab 95:3207–3215 [PubMed] [Google Scholar]490. Gibbs JP, Van Landingham C. 2008. Urinary perchlorate excretion does not predict thyroid function among pregnant women. Thyroid 18:807–808 [PubMed] [Google Scholar]491. Zoeller TR. 2010. Environmental chemicals targeting thyroid. Hormones 9:28–40 [PubMed] [Google Scholar]492. Fenner-Crisp PA. 2000. Endocrine modulators: risk characterization and assessment. Toxicol Pathol 28:438–440 [PubMed] [Google Scholar]493. Lucier GW. 1997. Dose-response relationships for endocrine disruptors: what we know and what we don’t know. Regul Toxicol Pharmacol 26:34–35 [PubMed] [Google Scholar]494. Sheehan DM, Willingham E, Gaylor D, Bergeron JM, Crews D. 1999. No threshold dose for estradiol-induced sex reversal of turtle embryos: how little is too much? Environ Health Perspect 107:155–159 [PMC free article] [PubMed] [Google Scholar]495. Sheehan DM, vom Saal FS. 1997. Low dose effects of hormones: a challenge for risk assessment. Risk Policy Report 4:31–39 [Google Scholar]496. Crews D, Bergeron JM, McLachlan JA. 1995. The role of estrogen in turtle sex determination and the effect of PCBs. Environ Health Perspect 103(Suppl 7):73–77 [PMC free article] [PubMed] [Google Scholar]497. vom Saal FS, Sheehan DM. 1998. Challenging risk assessment. Forum Appl Res Public Policy 13:11–18 [Google Scholar]498. Bergeron JM, Crews D, McLachlan JA. 1994. PCBs as environmental estrogens: turtle sex determination as a biomarker of environmental contamination. Environ Health Perspect 102:780–781 [PMC free article] [PubMed] [Google Scholar]499. Sonnenschein C, Olea N, Pasanen ME, Soto AM. 1989. Negative controls of cell proliferation: human prostate cancer cells and androgens. Cancer Res 49:3474–3481 [PubMed] [Google Scholar]500. Geck P, Szelei J, Jimenez J, Lin TM, Sonnenschein C, Soto AM. 1997. Expression of novel genes linked to the androgen-induced, proliferative shutoff in prostate cancer cells. J Steroid Biochem Mol Biol 63:211–218 [PubMed] [Google Scholar]501. Soto AM, Lin TM, Sakabe K, Olea N, Damassa DA, Sonnenschein C. 1995. Variants of the human prostate LNCaP cell line as a tool to study discrete components of the androgen-mediated proliferative response. Oncol Res 7:545–558 [PubMed] [Google Scholar]502. Geck P, Maffini MV, Szelei J, Sonnenschein C, Soto AM. 2000. Androgen-induced proliferative quiescence in prostate cancer: the role of AS3 as its mediator. Proc Natl Acad Sci USA 97:10185–10190 [PMC free article] [PubMed] [Google Scholar]503. Soto AM, Sonnenschein C. 1985. The role of estrogens on the proliferation of human breast tumor cells (MCF-7). J Steroid Biochem 23:87–94 [PubMed] [Google Scholar]504. Amara JF, Dannies PS. 1983. 17β-Estradiol has a biphasic effect on GH cell growth. Endocrinology 112:1141–1143 [PubMed] [Google Scholar]505. Soto AM, Sonnenschein C. 2001. The two faces of Janus: sex steroids as mediators of both cell proliferation and cell death. J Natl Cancer Inst 93:1673–1675 [PubMed] [Google Scholar]506. Sonnenschein C, Soto AM. 2008. Theories of carcinogenesis: an emerging perspective. Semin Cancer Biol 18:372–377 [PMC free article] [PubMed] [Google Scholar]507. Harris H. 2004. Tumour suppression: putting on the brakes. Nature 427:201. [PubMed] [Google Scholar]508. Yusuf I, Fruman DA. 2003. Regulation of quiescence in lymphocytes. Trends Immunol 24:380–386 [PubMed] [Google Scholar]509. Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. 2008. The ground state of embryonic stem cell self-renewal. Nature 453:519–523 [PMC free article] [PubMed] [Google Scholar]510. Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. 2006. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 38:1289–1297 [PubMed] [Google Scholar]511. Maffini M, Denes V, Sonnenschein C, Soto A, Geck P. 2008. APRIN is a unique Pds5 paralog with features of a chromatin regulator in hormonal differentiation. J Steroid Biochem Mol Biol 108:32–43 [PMC free article] [PubMed] [Google Scholar]512. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, Gustafsson JA. 2007. Estrogen receptors: how do they signal and what are their targets. Physiol Rev 87:905–931 [PubMed] [Google Scholar]513. Barkhem T, Nilsson S, Gustafsson JA. 2004. Molecular mechanisms, physiological consequences and pharmacological implications of estrogen receptor action. Am J Pharmacogenomics 4:19–28 [PubMed] [Google Scholar]514. Shi YB. 2009. Dual functions of thyroid hormone receptors in vertebrate development: the roles of histone-modifying cofactor complexes. Thyroid 19:987–999 [PMC free article] [PubMed] [Google Scholar]515. Kang HY, Tsai MY, Chang C, Huang KE. 2003. Mechanisms and clinical relevance of androgens and androgen receptor actions. Chang Gung Med J 26:388–402 [PubMed] [Google Scholar]516. Jeyakumar M, Webb P, Baxter JD, Scanlan TS, Katzenellenbogen JA. 2008. Quantification of ligand-regulated nuclear receptor corepressor and coactivator binding, key interactions determining ligand potency and efficacy for the thyroid hormone receptor. Biochemistry 47:7465–7476 [PMC free article] [PubMed] [Google Scholar]517. Nandi S. 1958. Endocrine control of mammary gland development and function in the C3H/ He Crgl mouse. J Natl Cancer Inst 21:1039–1063 [PubMed] [Google Scholar]518. Humphreys RC, Krajewska M, Krnacik S, Jaeger R, Weiher H, Krajewski S, Reed JC, Rosen JM. 1996. Apoptosis in the terminal end bud of the murine mammary gland: a mechanism of ductal morphogenesis. Development 122:4013–4022 [PubMed] [Google Scholar]519. Haslam SZ. 1986. Mammary fibroblast influence on normal mouse mammary epithelial cell responses to estrogen in vitro. Cancer Res 46:310–316 [PubMed] [Google Scholar]520. McGrath CM. 1983. Augmentation of the response of normal mammary epithelial cells to estradiol by mammary stroma. Cancer Res 43:1355–1360 [PubMed] [Google Scholar]521. Sohoni P, Sumpter JP. 1998. Several environmental oestrogens are also anti-androgens. J Endocrinol 158:327–339 [PubMed] [Google Scholar]522. Tilghman SL, Nierth-Simpson EN, Wallace R, Burow ME, McLachlan JA. 2010. Environmental hormones: Multiple pathways for response may lead to multiple disease outcomes. Steroids 75:520–523 [PubMed] [Google Scholar]523. Ismail A, Nawaz Z. 2005. Nuclear hormone receptor degradation and gene transcription: an update. IUBMB Life 57:483–490 [PubMed] [Google Scholar]524. Hoeck W, Rusconi S, Groner B. 1989. Down-regulation and phosphorylation of glucocorticoid receptors in cultured cells. Investigations with a monospecific antiserum against a bacterially expressed receptor fragment. J Biol Chem 264:14396–14402 [PubMed] [Google Scholar]525. Lange CA, Shen T, Horwitz KB. 2000. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA 97:1032–1037 [PMC free article] [PubMed] [Google Scholar]526. Nawaz Z, Lonard DM, Dennis AP, Smith CL, O’Malley BW. 1999. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci USA 96:1858–1862 [PMC free article] [PubMed] [Google Scholar]527. Lin HK, Altuwaijri S, Lin WJ, Kan PY, Collins LL, Chang C. 2002. Proteasome activity is required for androgen receptor transcriptional activity via regulation of androgen receptor nuclear translocation and interaction with coregulators in prostate cancer cells. J Biol Chem 277:36570–36576 [PubMed] [Google Scholar]528. von Zastrow M, Kobilka BK. 1994. Antagonist-dependent and -independent steps in the mechanism of adrenergic receptor internalization. J Biol Chem 269:18448–18452 [PubMed] [Google Scholar]529. Modrall JG, Nanamori M, Sadoshima J, Barnhart DC, Stanley JC, Neubig RR. 2001. ANG II type 1 receptor downregulation does not require receptor endocytosis or G protein coupling. Am J Physiol Cell Physiol 281:C801–C809 [PubMed] [Google Scholar]530. Kinyamu HK, Archer TK. 2003. Estrogen receptor-dependent proteasomal degradation of the glucocorticoid receptor is coupled to an increase in mdm2 protein expression. Mol Cell Biol 23:5867–5881 [PMC free article] [PubMed] [Google Scholar]531. Freedman NJ, Lefkowitz RJ. 1996. Desensitization of G protein-coupled receptors. Recent Prog Horm Res 51:319–351; discussion 352–353 [PubMed] [Google Scholar]532. Lohse MJ. 1993. Molecular mechanisms of membrane receptor desensitization. Biochim Biophys Acta 1179:171–188 [PubMed] [Google Scholar]533. Bohm SK, Grady EF, Bunnett NW. 1997. Regulatory mechanisms that modulate signalling by G-protein-coupled receptors. Biochem J 322:1–18 [PMC free article] [PubMed] [Google Scholar]534. Shankaran H, Wiley HS, Resat H. 2007. Receptor downregulation and desensitization enhance the information processing ability of signalling receptors. BMC Syst Biol 1:48. [PMC free article] [PubMed] [Google Scholar]535. Lesser B, Bruchovsky N. 1974. Effect of duration of the period after castration on the response of the rat ventral prostate to androgens. Biochem J 142:429–431 [PMC free article] [PubMed] [Google Scholar]536. Stormshak F, Leake R, Wertz N, Gorski J. 1976. Stimulatory and inhibitory effects of estrogen on uterine DNA synthesis. Endocrinology 99:1501–1511 [PubMed] [Google Scholar]537. Bruchovsky N, Lesser B, Van Doorn E, Craven S. 1975. Hormonal effects on cell proliferation in rat prostate. Vitam Horm 33:61–102 [PubMed] [Google Scholar]538. Coser KR, Chesnes J, Hur J, Ray S, Isselbacher KJ, Shioda T. 2003. Global analysis of ligand sensitivity of estrogen inducible and suppressible genes in MCF7/BUS breast cancer cells by DNA microarray. Proc Natl Acad Sci USA 100:13994–13999 [PMC free article] [PubMed] [Google Scholar]539. Hur J, Chesnes J, Coser KR, Lee RS, Geck P, Isselbacher KJ, Shioda T. 2004. The Bik BH3-only protein is induced in estrogen-starved and antiestrogen-exposed breast cancer cells and provokes apoptosis. Proc Natl Acad Sci USA 101:2351–2356 [PMC free article] [PubMed] [Google Scholar]540. Li L, Andersen ME, Heber S, Zhang Q. 2007. Non-monotonic dose-response relationship in steroid hormone receptor-mediated gene expression. J Mol Endocrinol 38:569–585 [PubMed] [Google Scholar]541. Vandenberg LN, Wadia PR, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. 2006. The mammary gland response to estradiol: monotonic at the cellular level, non-monotonic at the tissue-level of organization? J Steroid Biochem Mol Biol 101:263–274 [PubMed] [Google Scholar]542. Schell LM, Burnitz KK, Lathrop PW. 2010. Pollution and human biology. Ann Hum Biol 37:347–366 [PubMed] [Google Scholar]543. Plotkin D, Lechner JJ, Jung WE, Rosen PJ. 1978. Tamoxifen flare in advanced breast cancer. JAMA 240:2644–2646 [PubMed] [Google Scholar]544. Osborne CK, Hobbs K, Clark GM. 1985. Effect of estrogens and antiestrogens on growth of human breast cancer cells in athymic nude mice. Cancer Res 45:584–590 [PubMed] [Google Scholar]545. Berthois Y, Pons M, Dussert C, Crastes de Paulet A, Martin PM. 1994. Agonist-antagonist activity of anti-estrogens in the human breast cancer cell line MCF-7: an hypothesis for the interaction with a site distinct from the estrogen binding site. Mol Cell Endocrinol 99:259–268 [PubMed] [Google Scholar]546. Reddel RR, Sutherland RL. 1984. Tamoxifen stimulation of human breast cancer cell proliferation in vitro: a possible model for tamoxifen tumour flare. Eur J Cancer Clin Oncol 20:1419–1424 [PubMed] [Google Scholar]547. Wolf DM, Langan-Fahey SM, Parker CJ, McCague R, Jordan VC. 1993. Investigation of the mechanism of tamoxifen-stimulated breast tumor growth with nonisomerizable analogues of tamoxifen and metabolites. J Natl Cancer Inst 85:806–812 [PubMed] [Google Scholar]548. Howell A. 2001. Preliminary experience with pure antiestrogens. Clin Cancer Res 7:4369s–4375s; discusion 4411s–4412s [PubMed] [Google Scholar]549. Hattar R, Maller O, McDaniel S, Hansen KC, Hedman KJ, Lyons TR, Lucia S, Wilson RS, Jr, Schedin P. 2009. Tamoxifen induces pleiotrophic changes in mammary stroma resulting in extracellular matrix that suppresses transformed phenotypes. Breast Cancer Res 11:R5. [PMC free article] [PubMed] [Google Scholar]550. Howell A, Landberg G, Bergh J. 2009. Breast tumour stroma is a prognostic indicator and target for therapy. Breast Cancer Res 11(Suppl 3):S16. [PMC free article] [PubMed] [Google Scholar]551. Langan-Fahey SM, Tormey DC, Jordan VC. 1990. Tamoxifen metabolites in patients on long-term adjuvant therapy for breast cancer. Eur J Cancer 26:883–888 [PubMed] [Google Scholar]552. Kuiper GG, van den Bemd GJ, van Leeuwen JP. 1999. Estrogen receptor and the SERM concept. J Endocrinol Invest 22:594–603 [PubMed] [Google Scholar]553. MacGregor JI, Jordan VC. 1998. Basic guide to the mechanisms of antiestrogen action. Pharmacol Rev 50:151–196 [PubMed] [Google Scholar]554. Grese TA, Dodge JA. 1998. Selective estrogen receptor modulators (SERMs). Curr Pharm Des 4:71–92 [PubMed] [Google Scholar]555. Nagel SC, Hagelbarger JL, McDonnell DP. 2001. Development of an ER action indicator mouse for the study of estrogens, selective ER modulators (SERMs), and xenobiotics. Endocrinology 142:4721–4728 [PubMed] [Google Scholar]556. Gaido KW, Leonard LS, Lovell S, Gould JC, Babaï D, Portier CJ, McDonnell DP. 1997. Evaluation of chemicals with endocrine modulating activity in a yeast-based steroid hormone receptor gene transcription assay. Toxicol Appl Pharmacol 143:205–212 [PubMed] [Google Scholar]557. Gould JC, Leonard LS, Maness SC, Wagner BL, Conner K, Zacharewski T, Safe S, McDonnell DP, Gaido KW. 1998. Bisphenol A interacts with the estrogen receptor α in a distinct manner from estradiol. Mol Cell Endocrinol 142:203–214 [PubMed] [Google Scholar]558. Lerner HJ, Band PR, Israel L, Leung BS. 1976. Phase II study of tamoxifen: report of 74 patients with stage IV breast cancer. Cancer Treat Rep 60:1431–1435 [PubMed] [Google Scholar]559. Zhang HH, Kumar S, Barnett AH, Eggo MC. 1999. Intrinsic site-specific differences in the expression of leptin in human adipocytes and its autocrine effects on glucose uptake. J Clin Endocrinol Metab 84:2550–2556 [PubMed] [Google Scholar]560. Haddad N, Howland R, Baroody G, Daher C. 2006. The modulatory effect of leptin on the overall insulin production in ex-vivo normal rat pancreas. Can J Physiol Pharmacol 84:157–162 [PubMed] [Google Scholar]561. Pallett AL, Morton NM, Cawthorne MA, Emilsson V. 1997. Leptin inhibits insulin secretion and reduces insulin mRNA levels in rat isolated pancreatic islets. Biochem Biophys Res Commun 238:267–270 [PubMed] [Google Scholar]562. Thorburn AW, Holdsworth A, Proietto J, Morahan G. 2000. Differential and genetically separable associations of leptin with obesity-related traits. Int J Obes Relat Metab Disord 24:742–750 [PubMed] [Google Scholar]563. Lieb W, Sullivan LM, Harris TB, Roubenoff R, Benjamin EJ, Levy D, Fox CS, Wang TJ, Wilson PW, Kannel WB, Vasan RS. 2009. Plasma leptin levels and incidence of heart failure, cardiovascular disease, and total mortality in elderly individuals. Diabetes Care 32:612–616 [PMC free article] [PubMed] [Google Scholar]564. Neel BA, Sargis RM. 2011. The paradox of progress: environmental disruption of metabolism and the diabetes epidemic. Diabetes 60:1838–1848 [PMC free article] [PubMed] [Google Scholar]565. Sargis RM, Johnson DN, Choudhury RA, Brady MJ. 2010. Environmental endocrine disruptors promote adipogenesis in the 3T3-L1 cell line through glucocorticoid receptor activation. Obesity (Silver Spring) 18:1283–1288 [PMC free article] [PubMed] [Google Scholar]566. Hugo ER, Brandebourg TD, Woo JG, Loftus J, Alexander JW, Ben-Jonathan N. 2008. Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ Health Perspect 116:1642–1647 [PMC free article] [PubMed] [Google Scholar]567. Ben-Jonathan N, Hugo ER, Brandebourg TD. 2009. Effects of bisphenol A on adipokine release from human adipose tissue: implications for the metabolic syndrome. Mol Cell Endocrinol 304:49–54 [PMC free article] [PubMed] [Google Scholar]568. Miyawaki J, Sakayama K, Kato H, Yamamoto H, Masuno H. 2007. Perinatal and postnatal exposure to bisphenol a increases adipose tissue mass and serum cholesterol level in mice. J Atheroscler Thromb 14:245–252 [PubMed] [Google Scholar]569. Botelho GG, Golin M, Bufalo AC, Morais RN, Dalsenter PR, Martino-Andrade AJ. 2009. Reproductive effects of di(2-ethylhexyl)phthalate in immature male rats and its relation to cholesterol, testosterone, and thyroxin levels. Arch Environ Contam Toxicol 57:777–784 [PubMed] [Google Scholar]570. Lutz WK, Gaylor DW, Conolly RB, Lutz RW. 2005. Nonlinearity and thresholds in dose-response relationships for carcinogenicity due to sampling variation, logarithmic dose scaling, or small differences in individual susceptibility. Toxicol Appl Pharmacol 207:565–569 [PubMed] [Google Scholar]571. Center for the Evaluation of Risks to Human Reproduction 2007. NTP-CERHR expert panel report on the reproductive and developmental toxicity of bisphenol A. Washington, DC: Department of Health and Human Services [Google Scholar]572. Willhite CC, Ball GL, McLellan CJ. 2008. Derivation of a Bisphenol A organ reference dose (RfD) and drinking-water equivalent concentration. J Toxicol Environ Health B Crit Rev 11:69–146 [PubMed] [Google Scholar]573. Sakamoto H, Yokota H, Kibe R, Sayama Y, Yuasa A. 2002. Excretion of bisphenol A-glucuronide into the small intestine and deconjugation in the cecum of the rat. Biochem Biophys Acta 1573:171–176 [PubMed] [Google Scholar]574. Zalko D, Soto AM, Dolo L, Dorio C, Rathahao E, Debrauwer L, Faure R, Cravedi JP. 2003. Biotransformations of bisphenol A in a mammalian model: answers and new questions raised by low-dose metabolic fate studies in pregnant CD1 mice. Environ Health Perspect 111:309–319 [PMC free article] [PubMed] [Google Scholar]575. Stowell CL, Barvian KK, Young PC, Bigsby RM, Verdugo DE, Bertozzi CR, Widlanski TS. 2006. A role for sulfation-desulfation in the uptake of bisphenol A into breast tumor cells. Chem Biol 13:891–897 [PubMed] [Google Scholar]576. Center for the Evaluation of Risks to Human Reproduction 2008. Bisphenol A: public comments. Washington, DC: Department of Health and Human Services [Google Scholar]577. Markey CM, Michaelson CL, Veson EC, Sonnenschein C, Soto AM. 2001. The mouse uterotrophic assay: a reevaluation of its validity in assessing the estrogenicity of bisphenol A. Environ Health Perspect 109:55–60 [PMC free article] [PubMed] [Google Scholar]578. Schönfelder G, Friedrich K, Paul M, Chahoud I. 2004. Developmental effects of prenatal exposure to bisphenol A on the uterus of rat offspring. Neoplasia 6:584–594 [PMC free article] [PubMed] [Google Scholar]579. Eskenazi B, Mocarelli P, Warner M, Needham L, Patterson DG, Jr, Samuels S, Turner W, Gerthoux PM, Brambilla P. 2004. Relationship of serum TCDD concentrations and age at exposure of female residents of Seveso, Italy. Environ Health Perspect 112:22–27 [PMC free article] [PubMed] [Google Scholar]580. Warner M, Eskenazi B, Mocarelli P, Gerthoux PM, Samuels S, Needham L, Patterson D, Brambilla P. 2002. Serum dioxin concentrations and breast cancer risk in the Seveso Women’s Health Study. Environ Health Perspect 110:625–628 [PMC free article] [PubMed] [Google Scholar]581. Eskenazi B, Mocarelli P, Warner M, Samuels S, Vercellini P, Olive D, Needham LL, Patterson DG, Jr, Brambilla P, Gavoni N, Casalini S, Panazza S, Turner W, Gerthoux PM. 2002. Serum dioxin concentrations and endometriosis: a cohort study in Seveso, Italy. Environ Health Perspect 110:629–634 [PMC free article] [PubMed] [Google Scholar]582. Eskenazi B, Warner M, Mocarelli P, Samuels S, Needham LL, Patterson DG, Jr, Lippman S, Vercellini P, Gerthoux PM, Brambilla P, Olive D. 2002. Serum dioxin concentrations and menstrual cycle characteristics. Am J Epidemiol 156:383–392 [PubMed] [Google Scholar]583. Robinson GW, Karpf AB, Kratochwil K. 1999. Regulation of mammary gland development by tissue interaction. J Mammary Gland Biol Neoplasia 4:9–19 [PubMed] [Google Scholar]584. Medina D, Sivaraman L, Hilsenbeck SG, Conneely O, Ginger M, Rosen J, Omalle BW. 2001. Mechanisms of hormonal prevention of breast cancer. Ann NY Acad Sci 952:23–35 [PubMed] [Google Scholar]585. Schulz KM, Molenda-Figueira HA, Sisk CL. 2009. Back to the future: the organizational-activational hypothesis adapted to puberty and adolescence. Horm Behav 55:597–604 [PMC free article] [PubMed] [Google Scholar]586. Schulz KM, Sisk CL. 2006. Pubertal hormones, the adolescent brain, and the maturation of social behaviors: lessons from the Syrian hamster. Mol Cell Endocrinol 254–255:120–126 [PubMed] [Google Scholar]587. Primus RJ, Kellogg CK. 1990. Gonadal hormones during puberty organize environment-related social interaction in the male rat. Horm Behav 24:311–323 [PubMed] [Google Scholar]588. Arase S, Ishii K, Igarashi K, Aisaki K, Yoshio Y, Matsushima A, Shimohigashi Y, Arima K, Kanno J, Sugimura Y. 2011. Endocrine disrupter bisphenol A increases in situ estrogen production in the mouse urogenital sinus. Biol Reprod 84:734–742 [PubMed] [Google Scholar]589. Lee DH, Steffes MW, Sjödin A, Jones RS, Needham LL, Jacobs DR., Jr 2010. Low dose of some persistent organic pollutants predicts type 2 diabetes: a nested case-control study. Environ Health Perspect 118:1235–1242 [PMC free article] [PubMed] [Google Scholar]590. Lee DH, Steffes MW, Sjödin A, Jones RS, Needham LL, Jacobs DR., Jr 2011. Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS ONE 6:e15977. [PMC free article] [PubMed] [Google Scholar]591. Shin JY, Choi YY, Jeon HS, Hwang JH, Kim SA, Kang JH, Chang YS, Jacobs DR, Jr, Park JY, Lee DH. 2010. Low-dose persistent organic pollutants increased telomere length in peripheral leukocytes of healthy Koreans. Mutagenesis 25:511–516 [PubMed] [Google Scholar]592. MacLusky NJ, Hajszan T, Leranth C. 2005. The environmental estrogen bisphenol A inhibits estradiol-induced hippocampal synaptogenesis. Environ Health Perspect 113:675–679 [PMC free article] [PubMed] [Google Scholar]593. Della Seta D, Minder I, Dessì-Fulgheri F, Farabollini F. 2005. Bisphenol-A exposure during pregnancy and lactation affects maternal behavior in rats. Brain Res Bull 65:255–260 [PubMed] [Google Scholar]594. Razzoli M, Valsecchi P, Palanza P. 2005. Chronic exposure to low doses bisphenol A interferes with pair-bonding and exploration in female Mongolian gerbils. Brain Res Bull 65:249–254 [PubMed] [Google Scholar]595. Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A. 2006. The estrogenic effect of bisphenol A disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environ Health Perspect 114:106–112 [PMC free article] [PubMed] [Google Scholar]596. Titus-Ernstoff L, Hatch EE, Hoover RN, Palmer J, Greenberg ER, Ricker W, Kaufman R, Noller K, Herbst AL, Colton T, Hartge P. 2001. Long-term cancer risk in women given diethylstilbestrol (DES) during pregnancy. Br J Cancer 84:126–133 [PMC free article] [PubMed] [Google Scholar]597. Calle EE, Mervis CA, Thun MJ, Rodriguez C, Wingo PA, Heath CW., Jr 1996. Diethylstilbestrol and risk of fatal breast cancer in a prospective cohort of US women. Am J Epidemiol 144:645–652 [PubMed] [Google Scholar]598. Small CM, DeCaro JJ, Terrell ML, Dominguez C, Cameron LL, Wirth J, Marcus M. 2009. Maternal exposure to a brominated flame retardant and genitourinary conditions in male offspring. Environ Health Perspect 117:1175–1179 [PMC free article] [PubMed] [Google Scholar]599. Goldberg JM, Falcone T. 1999. Effect of diethylstilbestrol on reproductive function. Fertil Steril 72:1–7 [PubMed] [Google Scholar]600. Hatch EE, Herbst AL, Hoover RN, Noller KL, Adam E, Kaufman RH, Palmer JR, Titus-Ernstoff L, Hyer M, Hartge P, Robboy SJ. 2001. Incidence of squamous neoplasia of the cervix and vagina in women exposed prenatally to diethylstilbestrol (United States). Cancer Causes Control 12:837–845 [PubMed] [Google Scholar]601. Terrell ML, Berzen AK, Small CM, Cameron LL, Wirth JJ, Marcus M. 2009. A cohort study of the association between secondary sex ratio and parental exposure to polybrominated biphenyl (PBB) and polychlorinated biphenyl (PCB). Environ Health 8:35. [PMC free article] [PubMed] [Google Scholar]602. Xu X, Dailey AB, Talbott EO, Ilacqua VA, Kearney G, Asal NR. 2010. Associations of serum concentrations of organochlorine pesticides with breast cancer and prostate cancer in U.S. adults. Environ Health Perspect 118:60–66 [PMC free article] [PubMed] [Google Scholar]603. Li DK, Zhou Z, Miao M, He Y, Qing D, Wu T, Wang J, Weng X, Ferber J, Herrinton LJ, Zhu Q, Gao E, Yuan W. 2010. Relationship between urine bisphenol-A level and declining male sexual function. J Androl 31:500–506 [PubMed] [Google Scholar]604. Lim JS, Lee DH, Jacobs DR., Jr 2008. Association of brominated flame retardants with diabetes and metabolic syndrome in the U.S. population, 2003–2004. Diabetes Care 31:1802–1807 [PMC free article] [PubMed] [Google Scholar]605. Giordano F, Abballe A, De Felip E, di Domenico A, Ferro F, Grammatico P, Ingelido AM, Marra V, Marrocco G, Vallasciani S, Figà-Talamanca I. 2010. Maternal exposures to endocrine disrupting chemicals and hypospadias in offspring. Birth Defects Res A Clin Mol Teratol 88:241–250 [PubMed] [Google Scholar]606. Wolff MS, Engel SM, Berkowitz GS, Ye X, Silva MJ, Zhu C, Wetmur J, Calafat AM. 2008. Prenatal phenol and phthalate exposures and birth outcomes. Environ Health Perspect 116:1092–1097 [PMC free article] [PubMed] [Google Scholar]607. Sunyer J, Garcia-Esteban R, Alvarez M, Guxens M, Goñi F, Basterrechea M, Vrijheid M, Guerra S, Antó JM. 2010. DDE in mothers’ blood during pregnancy and lower respiratory tract infections in their infants. Epidemiology 21:729–735 [PubMed] [Google Scholar]608. World Health Organization 2002. Global assessment of the state-of-the-science of endocrine disruptors. Geneva: World Health Organization [Google Scholar]609. Tyl RW. 2009. Basic exploratory research versus guideline-compliant studies used for hazard evaluation and risk assessment: bisphenol A as a case study. Environ Health Perspect 117:1644–1651 [PMC free article] [PubMed] [Google Scholar]610. Tyl RW. 2010. In honor of the Teratology Society’s 50th anniversary: the role of Teratology Society members in the development and evolution of in vivo developmental toxicity test guidelines. Birth Defects Res C Embryo Today 90:99–102 [PubMed] [Google Scholar]611. Rice C, Birnbaum LS, Cogliano J, Mahaffey K, Needham L, Rogan WJ, vom Saal FS. 2003. Exposure assessment for endocrine disruptors: some considerations in the design of studies. Environ Health Perspect 111:1683–1690 [PMC free article] [PubMed] [Google Scholar]612. Soto AM, Rubin BS, Sonnenschein C. 2009. Interpreting endocrine disruption from an integrative biology perspective. Mol Cell Endocrinol 304:3–7 [PMC free article] [PubMed] [Google Scholar]613. Heindel JJ. 2008. Animal models for probing the developmental basis of disease and dysfunction paradigm. Basic Clin Pharmacol Toxicol 102:76–81 [PubMed] [Google Scholar]614. Heindel JJ, vom Saal FS. 2009. Role of nutrition and environmental endocrine disrupting chemicals during the perinatal period on the aetiology of obesity. Mol Cell Endocrinol 304:90–96 [PubMed] [Google Scholar]615. Newbold RR, Padilla-Banks E, Jefferson WN, Heindel JJ. 2008. Effects of endocrine disruptors on obesity. Int J Androl 31:201–208 [PubMed] [Google Scholar]616. Boobis AR, Doe JE, Heinrich-Hirsch B, Meek ME, Munn S, Ruchirawat M, Schlatter J, Seed J, Vickers C. 2008. IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit Rev Toxicol 38:87–96 [PubMed] [Google Scholar]617. German Federal Institute for Risk Assessment (BfR) 2009. Establishment of assessment and decision criteria in human health risk assessment for substances with endocrine disrupting properties under the EU plan protection product regulation. Report of a workshop hosted at the German Federal Institute for Risk Assessment (BfR), Berlin, Germany, 2009 [Google Scholar]618. Lidsky TI, Schneider JS. 2006. Adverse effects of childhood lead poisoning: the clinical neuropsychological perspective. Environ Res 100:284–293 [PubMed] [Google Scholar]619. Sheehan DM. 2006. No-threshold dose-response curves for nongenotoxic chemicals: findings and application for risk assessment. Environ Res 100:93–99 [PubMed] [Google Scholar]620. Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. 2009. Endocrine-disrupting chemical: an Endocrine Society scientific statement. Endocr Rev 30:293–342 [PMC free article] [PubMed] [Google Scholar]621. American Society of Human Genetics; American Society for Reproductive Medicine; Endocrine Society; Genetics Society of America; Society for Developmental Biology; Society for Pediatric Urology; Society for the Study of Reproduction; Society for Gynecologic Investigation 2011. Assessing chemical risk: societies offer expertise. Science 331:1136 [PubMed] [Google Scholar]622. Tominaga T, Negishi T, Hirooka H, Miyachi A, Inoue A, Hayasaka I, Yoshikawa Y. 2006. Toxicokinetics of bisphenol A in rats, monkeys and chimpanzees by the LC-MS/MS method. Toxicology 226:208–217 [PubMed] [Google Scholar]623. Newbold RR. 2004. Lessons learned from perinatal exposure to diethylstilbestrol. Toxicol Appl Pharmacol 199:142–150 [PubMed] [Google Scholar]624. Taylor JA, Vom Saal FS, Welshons WV, Drury B, Rottinghaus G, Hunt PA, Toutain PL, Laffont CM, VandeVoort CA. 2011. Similarity of bisphenol A pharmacokinetics in rhesus monkeys and mice: relevance for human exposure. Environ Health Perspect 119:422–430 [PMC free article] [PubMed] [Google Scholar]625. Gies A, Heinzow B, Dieter HH, Heindel J. 2009. Bisphenol A workshop of the German Federal Government Agency: March 30–31, 2009. Work group report: public health issues of bisphenol A. Int J Hyg Environ Health 212:693–696 [PubMed] [Google Scholar]626. World Health Organization 2010. Joint FAO/WHO expert meeting to review toxicological and health aspects of bisphenol A. Geneva: World Health Organization [Google Scholar]627. Kortenkamp A. 2008. Low dose mixture effects of endocrine disrupters: implications for risk assessment and epidemiology. Int J Androl 31:233–240 [PubMed] [Google Scholar]628. Bergeron JM, Willingham E, Osborn CT, 3rd, Rhen T, Crews D. 1999. Developmental synergism of steroidal estrogens in sex determination. Environ Health Perspect 107:93–97 [PMC free article] [PubMed] [Google Scholar]629. Rajapakse N, Silva E, Kortenkamp A. 2002. Combining xenoestrogens at levels below individual no-observed-effect concentrations dramatically enhances steroid hormone activity. Environ Health Perspect 110:917–921 [PMC free article] [PubMed] [Google Scholar]630. Rajapakse N, Silva E, Scholze M, Kortenkamp A. 2004. Deviation from additivity with estrogenic mixtures containing 4-nonylphenol and 4-tert-octylphenol detected in the E-SCREEN assay. Environ Sci Technol 38:6343–6352 [PubMed] [Google Scholar]631. Kortenkamp A, Faust M, Scholze M, Backhaus T. 2007. Low-level exposure to multiple chemicals: reason for human health concerns? Environ Health Perspect 115(Suppl 1):106–114 [PMC free article] [PubMed] [Google Scholar]632. Silins I, Högberg J. 2011. Combined toxic exposures and human health: biomarkers of exposure and effect. Int J Environ Res Public Health 8:629–647 [PMC free article] [PubMed] [Google Scholar]633. Rudel RA, Gray JM, Engel CL, Rawsthorne TW, Dodson RE, Ackerman JM, Rizzo J, Nudelman JL, Brody JG. 2011. Food packaging and bisphenol A and bis(2-ethylhexyl) phthalate exposure: findings from a dietary intervention. Environ Health Perspect 119:914–920 [PMC free article] [PubMed] [Google Scholar]634. Ji K, Kho YL, Park Y, Choi K. 2010. Influence of a five-day vegetarian diet on urinary levels of antibiotics and phthalate metabolites: a pilot study with “Temple Stay” participants Environ Res 110:375–382 [PubMed] [Google Scholar]635. Carwile JL, Luu HT, Bassett LS, Driscoll DA, Yuan C, Chang JY, Ye X, Calafat AM, Michels KB. 2009. Polycarbonate bottle use and urinary bisphenol A concentrations. Environ Health Perspect 117:1368–1372 [PMC free article] [PubMed] [Google Scholar]636. Matsumoto A, Kunugita N, Kitagawa K, Isse T, Oyama T, Foureman GL, Morita M, Kawamoto T. 2003. Bisphenol A levels in human urine. Environ Health Perspect 111:101–104 [PMC free article] [PubMed] [Google Scholar]637. Kawagoshi Y, Fujita Y, Kishi I, Fukunaga I. 2003. Estrogenic chemicals and estrogenic activity in leachate from municipal waste landfill determined by yeast two-hybrid assay. J Environ Monit 5:269–274 [PubMed] [Google Scholar]638. Liao C, Kannan K. 2011. High levels of bisphenol a in paper currencies from several countries, and implications for dermal exposure. Environ Sci Technol 45:6761–6768 [PubMed] [Google Scholar]639. Lopez-Espinosa MJ, Granada A, Araque P, Molina-Molina JM, Puertollano MC, Rivas A, Fernández M, Cerrillo I, Olea-Serrano MF, López C, Olea N. 2007. Oestrogenicity of paper and cardboard extracts used as food containers. Food Addit Contam 24:95–102 [PubMed] [Google Scholar]640. Terasaki M, Shiraishi F, Fukazawa H, Makino M. 2007. Occurrence and estrogenicity of phenolics in paper-recycling process water: pollutants originating from thermal paper in waste paper. Environ Toxicol Chem 26:2356–2366 [PubMed] [Google Scholar]641. Carson R. 1962. Silent spring. Boston, MA: Houghton Mifflin [Google Scholar]642. Chung E, Genco MC, Megrelis L, Ruderman JV. 2011. Effects of bisphenol A and triclocarban on brain-specific expression of aromatase in early zebrafish embryos. Proc Natl Acad Sci USA 108:17732–17737 [PMC free article] [PubMed] [Google Scholar]643. Rhee JS, Kim BM, Lee CJ, Yoon YD, Lee YM, Lee JS. 2011. Bisphenol A modulates expression of sex differentiation genes in the self-fertilizing fish, Kryptolebias marmoratus. Aquat Toxicol 104:218–229 [PubMed] [Google Scholar]644. Hatef A, Alavi SM, Abdulfatah A, Fontaine P, Rodina M, Linhart O. 2012. Adverse effects of bisphenol A on reproductive physiology in male goldfish at environmentally relevant concentrations. Ecotoxicol Environ Saf 76:56–62 [PubMed] [Google Scholar]645. Bai Y, Zhang YH, Zhai LL, Li XY, Yang J, Hong YY. 2011. Estrogen receptor expression and vitellogenin synthesis induced in hepatocytes of male frogs Rana chensinensis exposed to bisphenol A. Zool Res 32:317–322 [PubMed] [Google Scholar]646. Levy G, Lutz I, Krüger A, Kloas W. 2004. Bisphenol A induces feminization in Xenopus laevis tadpoles. Environ Res 94:102–111 [PubMed] [Google Scholar]647. Stoker C, Rey F, Rodriguez H, Ramos JG, Sirosky P, Larriera A, Luque EH, Muñoz-de-Toro M. 2003. Sex reversal effects on Caiman latirostris exposed to environmentally relevant doses of the xenoestrogen bisphenol A. Gen Comp Endocrinol 133:287–296 [PubMed] [Google Scholar]648. Stoker C, Beldoménico PM, Bosquiazzo VL, Zayas MA, Rey F, Rodríguez H, Muñoz-de-Toro M, Luque EH. 2008. Developmental exposure to endocrine disruptor chemicals alters follicular dynamics and steroid levels in Caiman latirostris. Gen Comp Endocrinol 156:603–612 [PubMed] [Google Scholar]649. Crain DA, Guillette LJ, Jr, Rooney AA, Pickford DB. 1997. Alterations in steroidogenesis in alligators (Alligator mississippiensis) exposed naturally and experimentally to environmental contaminants. Environ Health Perspect 105:528–533 [PMC free article] [PubMed] [Google Scholar]650. Mukhi S, Patiño R. 2007. Effects of prolonged exposure to perchlorate on thyroid and reproductive function in zebrafish. Toxicol Sci 96:246–254 [PubMed] [Google Scholar]651. Mukhi S, Torres L, Patiño R. 2007. Effects of larval-juvenile treatment with perchlorate and co-treatment with thyroxine on zebrafish sex ratios. Gen Comp Endocrinol 150:486–494 [PubMed] [Google Scholar]652. Bernhardt RR, von Hippel FA, O’Hara TM. 2011. Chronic perchlorate exposure causes morphological abnormalities in developing stickleback. Environ Toxicol Chem 30:1468–1478 [PMC free article] [PubMed] [Google Scholar]653. Li W, Zha J, Yang L, Li Z, Wang Z. 2011. Regulation of iodothyronine deiodinases and sodium iodide symporter mRNA expression by perchlorate in larvae and adult Chinese rare minnow (Gobiocypris rarus). Marine Pollut Bull 63:350–355 [PubMed] [Google Scholar]654. Goleman WL, Urquidi LJ, Anderson TA, Smith EE, Kendall RJ, Carr JA. 2002. Environmentally relevant concentrations of ammonium perchlorate inhibit development and metamorphosis in Xenopus laevis. Environ Toxicol Chem 21:424–430 [PubMed] [Google Scholar]655. Ortiz-Santaliestra ME, Sparling DW. 2007. Alteration of larval development and metamorphosis by nitrate and perchlorate in southern leopard frogs (Rana sphenocephala). Arch Environ Contam Toxicol 53:639–646 [PubMed] [Google Scholar]656. Hornung MW, Degitz SJ, Korte LM, Olson JM, Kosian PA, Linnum AL, Tietge JE. 2010. Inhibition of thyroid hormone release from cultured amphibian thyroid glands by methimazole, 6-propylthiouracil, and perchlorate. Toxicol Sci 118:42–51 [PubMed] [Google Scholar]657. Opitz R, Kloas W. 2010. Developmental regulation of gene expression in the thyroid gland of Xenopus laevis tadpoles. Gen Comp Endocrinol 168:199–208 [PubMed] [Google Scholar]658. Tietge JE, Butterworth BC, Haselman JT, Holcombe GW, Hornung MW, Korte JJ, Kosian PA, Wolfe M, Degitz SJ. 2010. Early temporal effects of three thyroid hormone synthesis inhibitors in Xenopus laevis. Aquat Toxicol 98:44–50 [PubMed] [Google Scholar]659. Chen Y, Sible JC, McNabb FMA. 2008. Effects of maternal exposure to ammonium perchlorate on thyroid function and the expression of thyroid-responsive genes in Japanese quail embryos. Gen Comp Endocrinol 159:196–207 [PubMed] [Google Scholar]660. Chen Y, McNabb FM, Sible JC. 2009. Perchlorate exposure induces hypothyroidism and affects thyroid-responsive genes in liver but not brain of quail chicks. Arch Environ Contam Toxicol 57:598–607 [PubMed] [Google Scholar]661. Pflugfelder O. 1959. The alteration of the thyroid and other organs of the domestic fowl by potassium perchlorate, with comparative studies on lower vertebrates. Wilhelm Roux Arch Entwicklungsmech Organ 151:78–112 [Google Scholar]662. Dent JN, Lynn WG. 1958. A comparison of the effects of goitrogens on thyroid activity in Triturus viridescens and Desmognathus fuscus. Biol Bull 115:411–420 [Google Scholar]663. Fox GA. 2001. Wildlife as sentinels of human health effects in the Great Lakes–St. Lawrence basin. Environ Health Perspect 109(Suppl 6):853–861 [PMC free article] [PubMed] [Google Scholar]664. Tanabe S. 2002. Contamination and toxic effects of persistent endocrine disrupters in marine mammals and birds. Mar Pollut Bull 45:69–77 [PubMed] [Google Scholar]665. Carney SA, Prasch AL, Heideman W, Peterson RE. 2006. Understanding dioxin developmental toxicity using the zebrafish model. Birth Defects Res A Clin Mol Teratol 76:7–18 [PubMed] [Google Scholar]666. Fisk AT, de Wit CA, Wayland M, Kuzyk ZZ, Burgess N, Letcher R, Braune B, Norstrom R, Blum SP, Sandau C, Lie E, Larsen HJ, Skaare JU, Muir DC. 2005. An assessment of the toxicological significance of anthropogenic contaminants in Canadian arctic wildlife. Sci Total Environ 351–352:57–93 [PubMed] [Google Scholar]667. Cooper KR, Wintermyer M. 2009. A critical review: 2,3,7,8-tetrachlorodibenzo-p-dioxin (2,3,7,8-TCDD) effects on gonad development in bivalve mollusks. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 27:226–245 [PubMed] [Google Scholar]668. Van den Berg M, Birnbaum L, Bosveld AT, Brunström B, Cook P, Feeley M, Giesy JP, Hanberg A, Hasegawa R, Kennedy SW, Kubiak T, Larsen JC, van Leeuwen FX, Liem AK, Nolt C, Peterson RE, Poellinger L, Safe S, Schrenk D, Tillitt D, Tysklind M, Younes M, Waern F, Zacharewski T. 1998. Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environ Health Perspect 106:775–792 [PMC free article] [PubMed] [Google Scholar]669. Gray LE, Ostby J, Wolf C, Lambright C, Kelce W. 1998. The value of mechanistic studies in laboratory animals for the prediction of reproductive effects in wildlife: endocrine effects on mammalian sexual differentiation. Environ Toxicol Chem 17:109–118 [Google Scholar]670. Hayes TB. 1998. Endocrine disruptors in amphibians: potential impacts and the usefulness of amphibian screens for detecting endocrine disrupting compounds. Sci J (Kagaku) 68:557–568 [Google Scholar]671. Colborn T. 1994. The wildlife/human connection: modernizing risk decisions. Environ Health Perspect 102:55–59 [PMC free article] [PubMed] [Google Scholar]672. Colborn T. 1995. Environmental estrogens: health implications for humans and wildlife. Environ Health Perspect 103:135–136 [PMC free article] [PubMed] [Google Scholar]673. Harrison PT, Holmes P, Humfrey CD. 1997. Reproductive health in humans and wildlife: are adverse trends associated with environmental chemical exposure? Sci Total Environ 205:97–106 [PubMed] [Google Scholar]674. Edwards TM, Moore BC, Guillette LJ., Jr 2006. Reproductive dysgenesis in wildlife: a comparative view. Int J Androl 29:109–121 [PubMed] [Google Scholar]675. Rhind SM. 2009. Anthropogenic pollutants: a threat to ecosystem sustainability? Philos Trans R Soc Lond B Biol Sci 364:3391–3401 [PMC free article] [PubMed] [Google Scholar]676. Decensi A, Gandini S, Guerrieri-Gonzaga A, Johansson H, Manetti L, Bonanni B, Sandri MT, Barreca A, Costa A, Robertson C, Lien EA. 1999. Effect of blood tamoxifen concentrations on surrogate biomarkers in a trial of dose reduction in healthy women. J Clin Oncol 17:2633–2638 [PubMed] [Google Scholar]677. Kisanga ER, Gjerde J, Guerrieri-Gonzaga A, Pigatto F, Pesci-Feltri A, Robertson C, Serrano D, Pelosi G, Decensi A, Lien EA. 2004. Tamoxifen and metabolite concentrations in serum and breast cancer tissue during three dose regimens in a randomized preoperative trial. Clin Cancer Res 10:2336–2343 [PubMed] [Google Scholar]678. Nagel SC, vom Saal FS, Welshons WV. 1998. The effective free fraction of estradiol and xenoestrogens in human serum measured by whole cell uptake assays: physiology of delivery modifies estrogenic activity. Proc Soc Exp Biol Med 217:300–309 [PubMed] [Google Scholar]679. Lakind JS, Naiman DQ. 2008. Bisphenol A (BPA) daily intakes in the United States: estimates from the 2003–2004 NHANES urinary BPA data. J Expo Sci Environ Epidemiol 18:608–615 [PubMed] [Google Scholar]680. Wittassek M, Koch HM, Angerer J, Brüning T. 2011. Assessing exposure to phthalates: the human biomonitoring approach. Mol Nutr Food Res 55:7–31 [PubMed] [Google Scholar]681. David RM, Moore MR, Finney DC, Guest D. 2000. Chronic toxicity of di(2-ethylhexyl)phthalate in rats. Toxicol Sci 55:433–443 [PubMed] [Google Scholar]682. Agency for Toxic Substances and Diseases Registry 2011. Toxic substances portal: di(2-ethylhexyl)phthalate (DEHP). Atlanta, GA: Centers for Disease Control [Google Scholar]683. Dickerson SM, Cunningham SL, Patisaul HB, Woller MJ, Gore AC. 2011. Endocrine disruption of brain sexual differentiation by developmental PCB exposure. Endocrinology 152:581–594 [PMC free article] [PubMed] [Google Scholar]684. Salama J, Chakraborty TR, Ng L, Gore AC. 2003. Effects of polychlorinated biphenyls on estrogen receptor-β expression in the anteroventral periventricular nucleus. Environ Health Perspect 111:1278–1282 [PMC free article] [PubMed] [Google Scholar]685. Cassidy RA, Vorhees CV, Minnema DJ, Hastings L. 1994. The effects of chlordane exposure during pre- and postnatal periods at environmentally relevant levels on sex steroid-mediated behaviors and functions in the rat. Toxicol Appl Pharmacol 126:326–337 [PubMed] [Google Scholar]686. McMahon T, Halstead N, Johnson S, Raffel TR, Romansic JM, Crumrine PW, Boughton RK, Martin LB, Rohr JR. 2011. The fungicide chlorothalonil is nonlinearly associated with corticosterone levels, immunity, and mortality in amphibians. Environ Health Perspect 119:1098–1103 [PMC free article] [PubMed] [Google Scholar]687. Guo-Ross SX, Chambers JE, Meek EC, Carr RL. 2007. Altered muscarinic acetylcholine receptor subtype binding in neonatal rat brain following exposure to chlorpyrifos or methyl parathion. Toxicol Sci 100:118–127 [PubMed] [Google Scholar]688. Palanza P, Parmigiani S, Liu H, vom Saal FS. 1999. Prenatal exposure to low doses of the estrogenic chemicals diethylstilbestrol and o,p’-DDT alters aggressive behavior of male and female house mice. Pharmacol Biochem Behav 64:665–672 [PubMed] [Google Scholar]689. vom Saal FS, Timms BG, Montano MM, Palanza P, Thayer KA, Nagel SC, Dhar MD, Ganjam VK, Parmigiani S, Welshons WV. 1997. Prostate enlargement in mice due to fetal exposure to low doses of estradiol or diethylstilbestrol and opposite effects at high doses. Proc Natl Acad Sci USA 94:2056–2061 [PMC free article] [PubMed] [Google Scholar]690. Slikker W, Jr, Scallet AC, Doerge DR, Ferguson SA. 2001. Gender-based differences in rats after chronic dietary exposure to genistein. Int J Toxicol 20:175–179 [PubMed] [Google Scholar]691. Smialowicz RJ, Williams WC, Copeland CB, Harris MW, Overstreet D, Davis BJ, Chapin RE. 2001. The effects of perinatal/juvenile heptachlor exposure on adult immune and reproductive system function in rats. Toxicol Sci 61:164–175 [PubMed] [Google Scholar]692. Valkusz Z, Nagyéri G, Radács M, Ocskó T, Hausinger P, László M, László FA, Juhász A, Julesz J, Pálföldi R, Gálfi M. 2011. Further analysis of behavioral and endocrine consequences of chronic exposure of male Wistar rats to subtoxic doses of endocrine disruptor chlorobenzenes. Physiol Behav 103:421–430 [PubMed] [Google Scholar]693. Manfo FP, Chao WF, Moundipa PF, Pugeat M, Wang PS. 2011. Effects of maneb on testosterone release in male rats. Drug Chem Toxicol 34:120–128 [PubMed] [Google Scholar]694. Chapin RE, Harris MW, Davis BJ, Ward SM, Wilson RE, Mauney MA, Lockhart AC, Smialowicz RJ, Moser VC, Burka LT, Collins BJ. 1997. The effects of perinatal/juvenile methoxychlor exposure on adult rat nervous, immune, and reproductive system function. Fundam Appl Toxicol 40:138–157 [PubMed] [Google Scholar]695. White KL, Jr, Germolec DR, Booker CD, Hernendez DM, McCay JA, Delclos KB, Newbold RR, Weis C, Guo TL. 2005. Dietary methoxychlor exposure modulates splenic natural killer cell activity, antibody-forming cell response and phenotypic marker expression in F0 and F1 generations of Sprague Dawley rats. Toxicology 207:271–281 [PubMed] [Google Scholar]696. Faass O, Schlumpf M, Reolon S, Henseler M, Maerkel K, Durrer S, Lichtensteiger W. 2009. Female sexual behavior, estrous cycle and gene expression in sexually dimorphic brain regions after pre- and postnatal exposure to endocrine active UV filters. Neurotoxicology 30:249–260 [PubMed] [Google Scholar]697. Lemini C, Hernández A, Jaimez R, Franco Y, Avila ME, Castell A. 2004. Morphometric analysis of mice uteri treated with the preservatives methyl, ethyl, propyl, and butylparaben. Toxicol Ind Health 20:123–132 [PubMed] [Google Scholar]698. Damgaard IN, Jensen TK, Petersen JH, Skakkebaek NE, Toppari J, Main KM. 2008. Risk factors for congenital cryptorchidism in a prospective birth cohort study. PLoS ONE 3:e3051. [PMC free article] [PubMed] [Google Scholar]699. Laurenzana EM, Weis CC, Bryant CW, Newbold R, Delclos KB. 2002. Effect of dietary administration of genistein, nonylphenol or ethinyl estradiol on hepatic testosterone metabolism, cytochrome P-450 enzymes, and estrogen receptor α expression. Food Chem Toxicol 40:53–63 [PubMed] [Google Scholar]700. Tyl RW, Myers CB, Marr MC, Brine DR, Fail PA, Seely JC, Van Miller JP. 1999. Two-generation reproduction study with para-tert-octylphenol in rats. Regul Toxicol Pharmacol 30:81–95 [PubMed] [Google Scholar]701. Li E, Guo Y, Ning Q, Zhang S, Li D. 2011. Research for the effect of octylphenol on spermatogenesis and proteomic analysis in octylphenol-treated mice testes. Cell Biol Int 35:305–309 [PubMed] [Google Scholar]702. Timofeeva OA, Sanders D, Seemann K, Yang L, Hermanson D, Regenbogen S, Agoos S, Kallepalli A, Rastogi A, Braddy D, Wells C, Perraut C, Seidler FJ, Slotkin TA, Levin ED. 2008. Persistent behavioral alterations in rats neonatally exposed to low doses of the organophosphate pesticide, parathion. Brain Res Bull 77:404–411 [PMC free article] [PubMed] [Google Scholar]703. Kuriyama SN, Wanner A, Fidalgo-Neto AA, Talsness CE, Koerner W, Chahoud I. 2007. Developmental exposure to low-dose PBDE-99: tissue distribution and thyroid hormone levels. Toxicology 242:80–90 [PubMed] [Google Scholar]704. Tanaka T, Morita A, Kato M, Hirai T, Mizoue T, Terauchi Y, Watanabe S, Noda M. 2011. Congener-specific polychlorinated biphenyls and the prevalence of diabetes in the Saku Control Obesity Program (SCOP). Endocr J 58:589–596 [PubMed] [Google Scholar]705. Buckman AH, Fisk AT, Parrott JL, Solomon KR, Brown SB. 2007. PCBs can diminish the influence of temperature on thyroid indices in rainbow trout (Oncorhynchus mykiss). Aquat Toxicol 84:366–378 [PubMed] [Google Scholar]706. Jiang Y, Zhao J, Van Audekercke R, Dequeker J, Geusens P. 1996. Effects of low-dose long-term sodium fluoride preventive treatment on rat bone mass and biomechanical properties. Calcif Tissue Int 58:30–39 [PubMed] [Google Scholar]707. Kirchner S, Kieu T, Chow C, Casey S, Blumberg B. 2010. Prenatal exposure to the environmental obesogen tributyltin predisposes multipotent stem cells to become adipocytes. Mol Endocrinol 24:526–539 [PMC free article] [PubMed] [Google Scholar]708. Stoker TE, Gibson EK, Zorrilla LM. 2010. Triclosan exposure modulates estrogen-dependent responses in the female wistar rat. Toxicol Sci 117:45–53 [PubMed] [Google Scholar]709. Eustache F, Mondon F, Canivenc-Lavier MC, Lesaffre C, Fulla Y, Berges R, Cravedi JP, Vaiman D, Auger J. 2009. Chronic dietary exposure to a low-dose mixture of genistein and vinclozolin modifies the reproductive axis, testis transcriptome, and fertility. Environ Health Perspect 117:1272–1279 [PMC free article] [PubMed] [Google Scholar]710. Schlumpf M, Durrer S, Faass O, Ehnes C, Fuetsch M, Gaille C, Henseler M, Hofkamp L, Maerkel K, Reolon S, Timms B, Tresguerres JA, Lichtensteiger W. 2008. Developmental toxicity of UV filters and environmental exposure: a review. Int J Androl 31:144–151 [PubMed] [Google Scholar]711. Schlecht C, Klammer H, Wuttke W, Jarry H. 2006. A dose-response study on the estrogenic activity of benzophenone-2 on various endpoints in the serum, pituitary and uterus of female rats. Arch Toxicol 80:656–661 [PubMed] [Google Scholar]712. Sitarek K. 2001. Embryolethal and teratogenic effects of carbendazim in rats. Teratog Carcinog Mutagen 21:335–340 [PubMed] [Google Scholar]713. Higashihara N, Shiraishi K, Miyata K, Oshima Y, Minobe Y, Yamasaki K. 2007. Subacute oral toxicity study of bisphenol F based on the draft protocol for the “Enhanced OECD Test Guideline no. 407”. Arch Toxicol 81:825–832 [PubMed] [Google Scholar]714. Yamano Y, Ohyama K, Ohta M, Sano T, Ritani A, Shimada J, Ashida N, Yoshida E, Ikehara K, Morishima I. 2005. A novel spermatogenesis related factor-2 (SRF-2) gene expression affected by TCDD treatment. Endocr J 52:75–81 [PubMed] [Google Scholar]715. Ikeda M, Tamura M, Yamashita J, Suzuki C, Tomita T. 2005. Repeated in utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure affects male gonads in offspring, leading to sex ratio changes in F2 progeny. Toxicol Appl Pharmacol 206:351–355 [PubMed] [Google Scholar]716. Welshons WV, Nagel SC, Thayer KA, Judy BM, Vom Saal FS. 1999. Low-dose bioactivity of xenoestrogens in animals: fetal exposure to low doses of methoxychlor and other xenoestrogens increases adult prostate size in mice. Toxicol Ind Health 15:12–25 [PubMed] [Google Scholar]717. Christian M, Gillies G. 1999. Developing hypothalamic dopaminergic neurones as potential targets for environmental estrogens. J Endocrinol 160:R1–R6 [PubMed] [Google Scholar]718. Jeng YJ, Watson CS. 2011. Combinations of physiologic estrogens with xenoestrogens alter ERK phosphorylation profiles in rat pituitary cells. Environ Health Perspect 119:104–112 [PMC free article] [PubMed] [Google Scholar]719. Jeng YJ, Kochukov MY, Watson CS. 2009. Membrane estrogen receptor-α-mediated nongenomic actions of phytoestrogens in GH3/B6/F10 pituitary tumor cells. J Mol Signal 4:2. [PMC free article] [PubMed] [Google Scholar]720. Narita S, Goldblum RM, Watson CS, Brooks EG, Estes DM, Curran EM, Midoro-Horiuti T. 2007. Environmental estrogens induce mast cell degradulation and enhance IgE-mediated release of allergic mediators. Environ Health Perspect 115:48–52 [PMC free article] [PubMed] [Google Scholar]721. Somjen D, Kohen F, Jaffe A, Amir-Zaltsman Y, Knoll E, Stern N. 1998. Effects of gonadal steroids and their antagonists on DNA synthesis in human vascular cells. Hypertension 32:39–45 [PubMed] [Google Scholar]722. Devidze N, Fujimori K, Urade Y, Pfaff DW, Mong JA. 2010. Estradiol regulation of lipocalin-type prostaglandin D synthase promoter activity: evidence for direct and indirect mechanisms. Neurosci Lett 474:17–21 [PMC free article] [PubMed] [Google Scholar]723. Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, Khairova R, Zhou R, Yuan P, Machado-Vieira R, McEwen BS, Manji HK. 2009. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci USA 106:3543–3548 [PMC free article] [PubMed] [Google Scholar]724. Guillen C, Bartolomé A, Nevado C, Benito M. 2008. Biphasic effect of insulin on β cell apoptosis depending on glucose deprivation. FEBS Lett 582:3855–3860 [PubMed] [Google Scholar]725. Welsh TH, Jr, Kasson BG, Hsueh AJ. 1986. Direct biphasic modulation of gonadotropin-stimulated testicular androgen biosynthesis by prolactin. Biol Reprod 34:796–804 [PubMed] [Google Scholar]726. Sarkar PK. 2008. l-Triiodothyronine differentially and nongenomically regulates synaptosomal protein phosphorylation in adult rat brain cerebral cortex: role of calcium and calmodulin. Life Sci 82:920–927 [PubMed] [Google Scholar]727. Calvo RM, Obregon MJ. 2009. Tri-iodothyronine upregulates adipnutrin mRNA expression in rat and human adipocytes. Mol Cell Endocrinol 311:39–46 [PubMed] [Google Scholar]728. Leung LY, Kwong AK, Man AK, Woo NY. 2008. Direct actions of cortisol, thyroxine and growth hormone on IGF-I mRNA expression in sea bream hepatocytes. Comp Biochem Physiol A Mol Integr Physiol 151:705–710 [PubMed] [Google Scholar]729. Habauzit D, Boudot A, Kerdivel G, Flouriot G, Pakdel F. 2010. Development and validation of a test for environmental estrogens: checking xeno-estrogen activity by CXCL12 secretion in breast cancer cell lines (CXCL-test). Environ Toxicol 25:495–503 [PubMed] [Google Scholar]730. Boettcher M, Kosmehl T, Braunbeck T. 2011. Low-dose effects and biphasic effect profiles: Is trenbolone a genotoxicant? Mutat Res 723:152–157 [PubMed] [Google Scholar]731. Wetherill YB, Petre CE, Monk KR, Puga A, Knudsen KE. 2002. The xenoestrogen bisphenol A induces inappropriate androgen receptor activation and mitogenesis in prostatic adenocarcinoma cells. Mol Cancer Ther 1:515–524 [PubMed] [Google Scholar]732. Sandy EH, Yao J, Zheng S, Gogra AB, Chen H, Zheng H, Yormah TB, Zhang X, Zaray G, Ceccanti B, Choi MM. 2010. A comparative cytotoxicity study of isomeric alkylphthalates to metabolically variant bacteria. J Hazard Mater 182:631–639 [PubMed] [Google Scholar]733. Murono EP, Derk RC, de León JH. 1999. Biphasic effects of octylphenol on testosterone biosynthesis by cultured Leydig cells from neonatal rats. Reprod Toxicol 13:451–462 [PubMed] [Google Scholar]734. Benísek M, Bláha L, Hilscherová K. 2008. Interference of PAHs and their N-heterocyclic analogs with signaling of retinoids in vitro. Toxicol In Vitro 22:1909–1917 [PubMed] [Google Scholar]735. Beníšek M, Kubincová P, Bláha L, Hilscherová K. 2011. The effects of PAHs and N-PAHs on retinoid signaling and Oct-4 expression in vitro. Toxicol Lett 200:169–175 [PubMed] [Google Scholar]736. Evanson M, Van Der Kraak GJ. 2001. Stimulatory effects of selected PAHs on testosterone production in goldfish and rainbow trout and possible mechanisms of action. Comp Biochem Physiol C Toxicol Pharmacol 130:249–258 [PubMed] [Google Scholar]737. Chaube R, Mishra S, Singh RK. 2010. In vitro effects of lead nitrate on steroid profiles in the post-vitellogenic ovary of the catfish Heteropneustes fossilis. Toxicol In Vitro 24:1899–1904 [PubMed] [Google Scholar]738. Helmestam M, Stavreus-Evers A, Olovsson M. 2010. Cadmium chloride alters mRNA levels of angiogenesis related genes in primary human endometrial endothelial cells grown in vitro. Reprod Toxicol 30:370–376 [PubMed] [Google Scholar]739. Chen AC, Donovan SM. 2004. Genistein at a concentration present in soy infant formula inhibits Caco-2BBe cell proliferation by causing G2/M cell cycle arrest. J Nutr 134:1303–1308 [PubMed] [Google Scholar]740. El Touny LH, Banerjee PP. 2009. Identification of a biphasic role for genistein in the regulation of prostate cancer growth and metastasis. Cancer Res 69:3695–3703 [PMC free article] [PubMed] [Google Scholar]741. Guo JM, Xiao BX, Liu DH, Grant M, Zhang S, Lai YF, Guo YB, Liu Q. 2004. Biphasic effect of daidzein on cell growth of human colon cancer cells. Food Chem Toxicol 42:1641–1646 [PubMed] [Google Scholar]742. Wang H, Zhou H, Zou Y, Liu Q, Guo C, Gao G, Shao C, Gong Y. 2010. Resveratrol modulates angiogenesis through the GSK3β/β-catenin/TCF-dependent pathway in human endothelial cells. Biochem Pharmacol 80:1386–1395 [PubMed] [Google Scholar]743. Pedro M, Lourenço CF, Cidade H, Kijjoa A, Pinto M, Nascimento MS. 2006. Effects of natural prenylated flavones in the phenotypical ER (+) MCF-7 and ER (−) MDA-MB-231 human breast cancer cells. Toxicol Lett 164:24–36 [PubMed] [Google Scholar]744. Almstrup K, Fernández MF, Petersen JH, Olea N, Skakkebaek NE, Leffers H. 2002. Dual effects of phytoestrogens result in U-shaped dose-response curves. Environ Health Perspect 110:743–748 [PMC free article] [PubMed] [Google Scholar]745. Pinto B, Bertoli A, Noccioli C, Garritano S, Reali D, Pistelli L. 2008. Estradiol-antagonistic activity of phenolic compounds from leguminous plants. Phytother Res 22:362–366 [PubMed] [Google Scholar]746. Sanderson JT, Hordijk J, Denison MS, Springsteel MF, Nantz MH, van den Berg M. 2004. Induction and inhibition of aromatase (CYP19) activity by natural and synthetic flavonoid compounds in H295R human adrenocortical carcinoma cells. Toxicol Sci 82:70–79 [PubMed] [Google Scholar]747. Elattar TM, Virji AS. 2000. The inhibitory effect of curcumin, genistein, quercetin and cisplatin on the growth of oral cancer cells in vitro. Anticancer Res 20:1733–1738 [PubMed] [Google Scholar]748. Ahn NS, Hu H, Park JS, Park JS, Kim JS, An S, Kong G, Aruoma OI, Lee YS, Kang KS. 2005. Molecular mechanisms of the 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced inverted U-shaped dose responsiveness in anchorage independent growth and cell proliferation of human breast epithelial cells with stem cell characteristics. Mutat Res 579:189–199 [PubMed] [Google Scholar]749. Dickerson SM, Guevara E, Woller MJ, Gore AC. 2009. Cell death mechanisms in GT1–7 GnRH cells exposed to polychlorinated biphenyls PCB74, PCB118, PCB153. Toxicol Appl Pharmacol 237:237–245 [PMC free article] [PubMed] [Google Scholar]750. Campagna C, Ayotte P, Sirard MA, Arsenault G, Laforest JP, Bailey JL. 2007. Effect of an environmentally relevant metabolized organochlorine mixture on porcine cumulus-oocyte complexes. Reprod Toxicol 23:145–152 [PubMed] [Google Scholar]751. Gasnier C, Dumont C, Benachour N, Clair E, Chagnon MC, Séralini GE. 2009. Glyphosate-based herbicides are toxic and endocrine disruptors in human cell lines. Toxicology 262:184–191 [PubMed] [Google Scholar]752. Greenman SB, Rutten MJ, Fowler WM, Scheffler L, Shortridge LA, Brown B, Sheppard BC, Deveney KE, Deveney CW, Trunkey DD. 1997. Herbicide/pesticide effects on intestinal epithelial growth. Environ Res 75:85–93 [PubMed] [Google Scholar]753. Sreeramulu K, Liu R, Sharom FJ. 2007. Interaction of insecticides with mammalian P-glycoprotein and their effect on its transport function. Biochim Biophys Acta 1768:1750–1757 [PubMed] [Google Scholar]754. Asp V, Ullerås E, Lindström V, Bergström U, Oskarsson A, Brandt I. 2010. Biphasic hormonal responses to the adrenocorticolytic DDT metabolite 3-methylsulfonyl-DDE in human cells. Toxicol Appl Pharmacol 242:281–289 [PubMed] [Google Scholar]755. Ralph JL, Orgebin-Crist MC, Lareyre JJ, Nelson CC. 2003. Disruption of androgen regulation in the prostate by the environmental contaminant hexachlorobenzene. Environ Health Perspect 111:461–466 [PMC free article] [PubMed] [Google Scholar]756. Ohlsson A, Ullerås E, Oskarsson A. 2009. A biphasic effect of the fungicide prochloraz on aldosterone, but not cortisol, secretion in human adrenal H295R cells: underlying mechanisms. Toxicol Lett 191:174–180 [PubMed] [Google Scholar]757. Ohlsson A, Cedergreen N, Oskarsson A, Ullerås E. 2010. Mixture effects of imidazole fungicides on cortisol and aldosterone secretion in human adrenocortical H295R cells. Toxicology 275:21–28 [PubMed] [Google Scholar]758. Kim KH, Bose DD, Ghogha A, Riehl J, Zhang R, Barnhart CD, Lein PJ, Pessah IN. 2011. Para- and ortho-substitutions are key determinants of polybrominated diphenyl ether activity toward ryanodine receptors and neurotoxicity. Environ Health Perspect 119:519–526 [PMC free article] [PubMed] [Google Scholar]759. Alm H, Scholz B, Kultima K, Nilsson A, Andrén PE, Savitski MM, Bergman A, Stigson M, Fex-Svenningsen A, Dencker L. 2010. In vitro neurotoxicity of PBDE-99: immediate and concentration-dependent effects on protein expression in cerebral cortex cells. J Proteome Res 9:1226–1235 [PubMed] [Google Scholar]760. Sànchez JJ, Abreu P, González-Hernández T, Hernández A, Prieto L, Alonso R. 2004. Estrogen modulation of adrenoceptor responsiveness in the female rat pineal gland: differential expression of intracellular estrogen receptors. J Pineal Res 37:26–35 [PubMed] [Google Scholar]761. Shelby MD, Newbold RR, Tully DB, Chae K, Davis VL. 1996. Assessing environmental chemicals for estrogenicity using a combination of in vitro and in vivo assays. Environ Health Perspect 104:1296–1300 [PMC free article] [PubMed] [Google Scholar]762. Dhir A, Kulkarni SK. 2008. Antidepressant-like effect of 17β-estradiol: involvement of dopaminergic, serotonergic, and (or) sigma-1 receptor systems. Can J Physiol Pharmacol 86:726–735 [PubMed] [Google Scholar]763. Ribeiro AC, Pfaff DW, Devidze N. 2009. Estradiol modulates behavioral arousal and induces changes in gene expression profiles in brain regions involved in the control of vigilance. Eur J Neurosci 29:795–801 [PubMed] [Google Scholar]764. Park CR, Campbell AM, Woodson JC, Smith TP, Fleshner M, Diamond DM. 2006. Permissive influence of stress in the expression of a U-shaped relationship between serum corticosterone levels and spatial memory errors in rats. Dose Response 4:55–74 [PMC free article] [PubMed] [Google Scholar]765. Abrahám I, Harkany T, Horvath KM, Veenema AH, Penke B, Nyakas C, Luiten PG. 2000. Chronic corticosterone administration dose-dependently modulates Aβ(1–42)- and NMDA-induced neurodegeneration in rat magnocellular nucleus basalis. J Neuroendocrinol 12:486–494 [PubMed] [Google Scholar]766. Duclos M, Gouarne C, Martin C, Rocher C, Mormède P, Letellier T. 2004. Effects of corticosterone on muscle mitochondria identifying different sensitivity to glucocorticoids in Lewis and Fischer rats. Am J Physiol Endocrinol Metab 286:E159–E167 [PubMed] [Google Scholar]767. Abrari K, Rashidy-Pour A, Semnanian S, Fathollahi Y, Jadid M. 2009. Post-training administration of corticosterone enhances consolidation of contextual fear memory and hippocampal long-term potentiation in rats. Neurobiol Learn Mem 91:260–265 [PubMed] [Google Scholar]768. Spée M, Marchal L, Thierry AM, Chastel O, Enstipp M, Maho YL, Beaulieu M, Raclot T. 2011. Exogenous corticosterone mimics a late fasting stage in captive Adelie penguins (Pygoscelis adeliae). Am J Physiol Regul Integr Comp Physiol 300:R1241–R1249 [PubMed] [Google Scholar]769. Sunny F, Oommen VO. 2004. Effects of steroid hormones on total brain Na+-K+ ATPase activity in Oreochromis mossambicus. Indian J Exp Biol 42:283–287 [PubMed] [Google Scholar]770. Huggard D, Khakoo Z, Kassam G, Mahmoud SS, Habibi HR. 1996. Effect of testosterone on maturational gonadotropin subunit messenger ribonucleic acid levels in the goldfish pituitary. Biol Reprod 54:1184–1191 [PubMed] [Google Scholar]771. Ren SG, Huang Z, Sweet DE, Malozowski S, Cassorla F. 1990. Biphasic response of rat tibial growth to thyroxine administration. Acta Endocrinol (Copenh) 122:336–340 [PubMed] [Google Scholar]772. Houshmand F, Faghihi M, Zahediasl S. 2009. Biphasic protective effect of oxytocin on cardiac ischemia/reperfusion injury in anaesthetized rats. Peptides 30:2301–2308 [PubMed] [Google Scholar]773. Boccia MM, Kopf SR, Baratti CM. 1998. Effects of a single administration of oxytocin or vasopressin and their interactions with two selective receptor antagonists on memory storage in mice. Neurobiol Learn Mem 69:136–146 [PubMed] [Google Scholar]774. Tai SH, Hung YC, Lee EJ, Lee AC, Chen TY, Shen CC, Chen HY, Lee MY, Huang SY, Wu TS. 2011. Melatonin protects against transient focal cerebral ischemia in both reproductively active and estrogen-deficient female rats: the impact of circulating estrogen on its hormetic dose-response. J Pineal Res 50:292–303 [PubMed] [Google Scholar]775. Cai JX, Arnsten AF. 1997. Dose-dependent effects of the dopamine D1 receptor agonists A77636 or SKF81297 on spatial working memory in aged monkeys. J Pharmacol Exp Ther 283:183–189 [PubMed] [Google Scholar]776. Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. 2007. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci 10:376–384 [PubMed] [Google Scholar]777. Palanza P, Parmigiani S, vom Saal FS. 2001. Effects of prenatal exposure to low doses of diethylstilbestrol, o,p’DDT, and methoxychlor on postnatal growth and neurobehavioral development in male and female mice. Horm Behav 40:252–265 [PubMed] [Google Scholar]778. Thuillier R, Wang Y, Culty M. 2003. Prenatal exposure to estrogenic compounds alters the expression pattern of platelet-derived growth factor receptors α and β in neonatal rat testis: identification of gonocytes as targets of estrogen exposure. Biol Reprod 68:867–880 [PubMed] [Google Scholar]779. Köhlerová E, Skarda J. 2004. Mouse bioassay to assess oestrogenic and anti-oestrogenic compounds: hydroxytamoxifen, diethylstilbestrol and genistein. J Vet Med A Physiol Pathol Clin Med 51:209–217 [PubMed] [Google Scholar]780. Putz O, Schwartz CB, Kim S, LeBlanc GA, Cooper RL, Prins GS. 2001. Neonatal low- and high-dose exposure to estradiol benzoate in the male rat. I. Effects on the prostate gland. Biol Reprod 65:1496–1505 [PubMed] [Google Scholar]781. Rochester JR, Forstmeier W, Millam JR. 2010. Post-hatch oral estrogen in zebra finches (Taeniopygia guttata): is infertility due to disrupted testes morphology or reduced copulatory behavior? Physiol Behav 101:13–21 [PubMed] [Google Scholar]782. Vosges M, Le Page Y, Chung BC, Combarnous Y, Porcher JM, Kah O, Brion F. 2010. 17α-Ethinylestradiol disrupts the ontogeny of the forebrain GnRH system and the expression of brain aromatase during early development of zebrafish. Aquat Toxicol 99:479–491 [PubMed] [Google Scholar]783. Gust M, Buronfosse T, Giamberini L, Ramil M, Mons R, Garric J. 2009. Effects of fluoxetine on the reproduction of two prosobranch mollusks: Potamopyrgus antipodarum and Valvata piscinalis. Environ Pollut 157:423–429 [PubMed] [Google Scholar]784. Villeneuve DL, Knoebl I, Kahl MD, Jensen KM, Hammermeister DE, Greene KJ, Blake LS, Ankley GT. 2006. Relationship between brain and ovary aromatase activity and isoform-specific aromatase mRNA expression in the fathead minnow (Pimephales promelas). Aquat Toxicol 76:353–368 [PubMed] [Google Scholar]785. Jones BA, Shimell JJ, Watson NV. 2011. Pre- and postnatal Bisphenol A treatment results in persistent deficits in the sexual behavior of male rats, but not female rats, in adulthood. Horm Behav 59:246–251 [PubMed] [Google Scholar]786. Lemos MF, Esteves AC, Samyn B, Timperman I, van Beeumen J, Correia A, van Gestel CA, Soares AM. 2010. Protein differential expression induced by endocrine disrupting compounds in a terrestrial isopod. Chemosphere 79:570–576 [PubMed] [Google Scholar]787. Nishizawa H, Morita M, Sugimoto M, Imanishi S, Manabe N. 2005. Effects of in utero exposure to bisphenol A on mRNA expression of arylhydrocarbon and retinoid receptors in murine embryos. J Reprod Dev 51:315–324 [PubMed] [Google Scholar]788. Andrade AJ, Grande SW, Talsness CE, Grote K, Chahoud I. 2006. A dose-response study following in utero and lactational exposure to di-(2-ethylhexyl)-phthalate (DEPH): non-monotonic dose-response and low dose effects on rat brain aromatase activity. Toxicology 227:185–192 [PubMed] [Google Scholar]789. Ge RS, Chen GR, Dong Q, Akingbemi B, Sottas CM, Santos M, Sealfon SC, Bernard DJ, Hardy MP. 2007. Biphasic effects of postnatal exposure to diethylhexylphthalate on the timing of puberty in male rats. J Androl 28:513–520 [PubMed] [Google Scholar]790. Grande SW, Andrade AJ, Talsness CE, Grote K, Chahoud I. 2006. A dose-response study following in utero and lactational exposure to di(2-ethylhexyl)phthalate: effects on female rat reproductive development. Toxicol Sci 91:247–254 [PubMed] [Google Scholar]791. Vo TT, Jung EM, Dang VH, Yoo YM, Choi KC, Yu FH, Jeung EB. 2009. Di-(2 ethylhexyl) phthalate and flutamide alter gene expression in the testis of immature male rats. Reprod Biol Endocrinol 7:104. [PMC free article] [PubMed] [Google Scholar]792. Takano H, Yanagisawa R, Inoue K, Ichinose T, Sadakane K, Yoshikawa T. 2006. Di-(2-ethylhexyl) phthalate enhances atopic dermatitis-like skin lesions in mice. Environ Health Perspect 114:1266–1269 [PMC free article] [PubMed] [Google Scholar]793. Oliveira-Filho EC, Grisolia CK, Paumgartten FJR. 2009. Trans-generation study of the effects of nonylphenol ethoxylate on the reproduction of the snail Biomphalaria tenagophila. Ecotoxicol Environ Saf 72:458–465 [PubMed] [Google Scholar]794. Duft M, Schulte-Oehlmann U, Weltje L, Tillmann M, Oehlmann J. 2003. Stimulated embryo production as a parameter of estrogenic exposure via sediments in the freshwater mudsnail Potamopyrgus antipodarum. Aquat Toxicol 64:437–449 [PubMed] [Google Scholar]795. Oehlmann J, Schulte-Oehlmann U, Tillmann M, Markert B. 2000. Effects of endocrine disruptors on prosobranch snails (Mollusca: Gastropoda) in the laboratory. Part I. bisphenol A and octylphenol as xeno-estrogens. Ecotoxicology 9:383–397 [PubMed] [Google Scholar]796. Maranghi F, Tassinari R, Marcoccia D, Altieri I, Catone T, De Angelis G, Testai E, Mastrangelo S, Evandri MG, Bolle P, Lorenzetti S. 2010. The food contaminant semicarbazide acts as an endocrine disrupter: evidence from an integrated in vivo/in vitro approach. Chem Biol Interact 183:40–48 [PubMed] [Google Scholar]797. Giudice BD, Young TM. 2010. The antimicrobial triclocarban stimulates embryo production in the freshwater mudsnail Potamopyrgus antipodarum. Environ Toxicol Chem 29:966–970 [PMC free article] [PubMed] [Google Scholar]798. Love OP, Shutt LJ, Silfies JS, Bortolotti GR, Smits JE, Bird DM. 2003. Effects of dietary PCB exposure on adrenocortical function in captive American kestrels (Falco sparverius). Ecotoxicology 12:199–208 [PubMed] [Google Scholar]799. Franceschini MD, Custer CM, Custer TW, Reed JM, Romero LM. 2008. Corticosterone stress response in tree swallows nesting near polychlorinated biphenyl- and dioxin-contaminated rivers. Environ Toxicol Chem 27:2326–2331 [PubMed] [Google Scholar]800. Axelstad M, Boberg J, Hougaard KS, Christiansen S, Jacobsen PR, Mandrup KR, Nellemann C, Lund SP, Hass U. 2011. Effects of pre- and postnatal exposure to the UV-filter octyl methoxycinnamate (OMC) on the reproductive, auditory and neurological development of rat offspring. Toxicol Appl Pharmacol 250:278–290 [PubMed] [Google Scholar]801. Riegel AC, French ED. 1999. Acute toluene induces biphasic changes in rat spontaneous locomotor activity which are blocked by remoxipride. Pharmacol Biochem Behav 62:399–402 [PubMed] [Google Scholar]802. Fan F, Wierda D, Rozman KK. 1996. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on humoral and cell-mediated immunity in Sprague-Dawley rats. Toxicology 106:221–228 [PubMed] [Google Scholar]803. Teeguarden JG, Dragan YP, Singh J, Vaughan J, Xu YH, Goldsworthy T, Pitot HC. 1999. Quantitative analysis of dose- and time-dependent promotion of four phenotypes of altered hepatic foci by 2,3,7,8-tetrachlorodibenzo-p-dioxin in female Sprague-Dawley rats. Toxicol Sci 51:211–223 [PubMed] [Google Scholar]804. Höfer N, Diel P, Wittsiepe J, Wilhelm M, Kluxen FM, Degen GH. 2010. Investigations on the estrogenic activity of the metallohormone cadmium in the rat intestine. Arch Toxicol 84:541–552 [PubMed] [Google Scholar]805. Zhang Y, Shen G, Yu Y, Zhu H. 2009. The hormetic effect of cadmium on the activity of antioxidant enzymes in the earthworm Eisenia fetida. Environ Pollut 157:3064–3068 [PubMed] [Google Scholar]806. Sharma B, Patiño R. 2009. Effects of cadmium on growth, metamorphosis and gonadal sex differentiation in tadpoles of the African clawed frog, Xenopus laevis. Chemosphere 76:1048–1055 [PubMed] [Google Scholar]807. Wang CR, Tian Y, Wang XR, Yu HX, Lu XW, Wang C, Wang H. 2010. Hormesis effects and implicative application in assessment of lead-contaminated soils in roots of Vicia faba seedlings. Chemosphere 80:965–971 [PubMed] [Google Scholar]808. Fox DA, Kala SV, Hamilton WR, Johnson JE, O’Callaghan JP. 2008. Low-level human equivalent gestational lead exposure produces supernormal scotopic electroretinograms, increased retinal neurogenesis, and decreased retinal dopamine utilization in rats. Environ Health Perspect 116:618–625 [PMC free article] [PubMed] [Google Scholar]809. Chiang EC, Shen S, Kengeri SS, Xu H, Combs GF, Morris JS, Bostwick DG, Waters DJ. 2009. Defining the optimal selenium dose for prostate cancer risk reduction: insights from the U-shaped relationship between selenium status, DNA damage, and apoptosis. Dose Response 8:285–300 [PMC free article] [PubMed] [Google Scholar]810. Harding LE. 2008. Non-linear uptake and hormesis effects of selenium in red-winged blackbirds (Agelaius phoeniceus). Sci Total Environ 389:350–366 [PubMed] [Google Scholar]811. Wisniewski AB, Cernetich A, Gearhart JP, Klein SL. 2005. Perinatal exposure to genistein alters reproductive development and aggressive behavior in male mice. Physiol Behav 84:327–334 [PubMed] [Google Scholar]812. Anderson JJ, Ambrose WW, Garner SC. 1998. Biphasic effects of genistein on bone tissue in the ovariectomized, lactating rat model. Proc Soc Exp Biol Med 217:345–350 [PubMed] [Google Scholar]813. Dey A, Guha P, Chattopadhyay S, Bandyopadhyay SK. 2009. Biphasic activity of resveratrol on indomethacin-induced gastric ulcers. Biochem Biophys Res Commun 381:90–95 [PubMed] [Google Scholar]814. Boccia MM, Kopf SR, Baratti CM. 1999. Phlorizin, a competitive inhibitor of glucose transport, facilitates memory storage in mice. Neurobiol Learn Mem 71:104–112 [PubMed] [Google Scholar]815. Brodeur JC, Svartz G, Perez-Coll CS, Marino DJ, Herkovits J. 2009. Comparative susceptibility to atrazine of three developmental stages of Rhinella arenarum and influence on metamorphosis: non-monotonous acceleration of the time to climax and delayed tail resorption. Aquat Toxicol 91:161–170 [PubMed] [Google Scholar]816. Freeman JL, Beccue N, Rayburn AL. 2005. Differential metamorphosis alters the endocrine response in anuran larvae exposed to T3 and atrazine. Aquat Toxicol 75:263–276 [PubMed] [Google Scholar]817. Undeðer U, Schlumpf M, Lichtensteiger W. 2010. Effect of the herbicide pendimethalin on rat uterine weight and gene expression and in silico receptor binding analysis. Food Chem Toxicol 48:502–508 [PubMed] [Google Scholar]818. Cavieres MF, Jaeger J, Porter W. 2002. Developmental toxicity of a commercial herbicide mixture in mice. I. Effects on embryo implantation and litter size. Environ Health Perspect 110:1081–1085 [PMC free article] [PubMed] [Google Scholar]819. Zorrilla LM, Gibson EK, Stoker TE. 2010. The effects of simazine, a chlorotriazine herbicide, on pubertal development in the female Wistar rat. Reprod Toxicol 29:393–400 [PubMed] [Google Scholar]820. Bloomquist JR, Barlow RL, Gillette JS, Li W, Kirby ML. 2002. Selective effects of insecticides on nigrostriatal dopaminergic nerve pathways. Neurotoxicology 23:537–544 [PubMed] [Google Scholar]821. Lassiter TL, Brimijoin S. 2008. Rats gain excess weight after developmental exposure to the organophosphorothionate pesticide, chlorpyrifos. Neurotoxicol Teratol 30:125–130 [PubMed] [Google Scholar]822. Wu H, Zhang R, Liu J, Guo Y, Ma E. 2011. Effects of malathion and chlorpyrifos on acetylcholinesterase and antioxidant defense system in Oxya chinensis (Thunberg) (Orthoptera: Acrididae). Chemosphere 83:599–604 [PubMed] [Google Scholar]823. Muthuviveganandavel V, Muthuraman P, Muthu S, Srikumar K. 2008. Toxic effects of carbendazim at low dose levels in male rats. J Toxicol Sci 33:25–30 [PubMed] [Google Scholar]824. Laughlin GA, Goodell V, Barrett-Connor E. 2010. Extremes of endogenous testosterone are associated with increased risk of incident coronary events in older women. J Clin Endocrinol Metab 95:740–747 [PMC free article] [PubMed] [Google Scholar]825. Kratzik CW, Schatzl G, Lackner JE, Lunglmayr G, Brandstätter N, Rücklinger E, Huber J. 2007. Mood changes, body mass index and bioavailable testosterone in healthy men: results of the Androx Vienna Municipality Study. BJU Int 100:614–618 [PubMed] [Google Scholar]826. Floege J, Kim J, Ireland E, Chazot C, Drueke T, de Francisco A, Kronenberg F, Marcelli D, Passlick-Deetjen J, Schernthaner G, Fouqueray B, Wheeler DC. 2010. Serum iPTH, calcium and phosphate, and the risk of mortality in a European haemodialysis population. Nephrol Dial Transplant 26:1948–1955 [PMC free article] [PubMed] [Google Scholar]827. Danese MD, Kim J, Doan QV, Dylan M, Griffiths R, Chertow GM. 2006. PTH and the risks for hip, vertebral, and pelvic fractures among patients on dialysis. Am J Kidney Dis 47:149–156 [PubMed] [Google Scholar]828. Tan ZS, Beiser A, Vasan RS, Au R, Auerbach S, Kiel DP, Wolf PA, Seshadri S. 2008. Thyroid function and the risk of Alzheimer disease: the Framingham Study. Arch Intern Med 168:1514–1520 [PMC free article] [PubMed] [Google Scholar]829. Tanaka M, Fukui M, Tomiyasu K, Akabame S, Nakano K, Hasegawa G, Oda Y, Nakamura N. 2010. U-shaped relationship between insulin level and coronary artery calcification (CAC). J Atheroscler Thromb 17:1033–1040 [PubMed] [Google Scholar]830. Pyörälä M, Miettinen H, Laakso M, Pyörälä K. 2000. Plasma insulin and all-cause, cardiovascular, and noncardiovascular mortality: the 22-year follow-up results of the Helsinki Policemen Study. Diabetes Care 23:1097–1102 [PubMed] [Google Scholar]831. Kumari M, Chandola T, Brunner E, Kivimaki M. 2010. A nonlinear relationship of generalized and central obesity with diurnal cortisol secretion in the Whitehall II study. J Clin Endocrinol Metab 95:4415–4423 [PMC free article] [PubMed] [Google Scholar]832. Bremmer MA, Deeg DJ, Beekman AT, Penninx BW, Lips P, Hoogendijk WJ. 2007. Major depression in late life is associated with both hypo- and hypercortisolemia. Biol Psychiatry 62:479–486 [PubMed] [Google Scholar]833. Lee DH, Steffes MW, Sjödin A, Jones RS, Needham LL, Jacobs DR., Jr 2010. Low dose of some persistent organic pollutants predicts type 2 diabetes: a nested case-control study. Environ Health Perspect 118:1235–1242 [PMC free article] [PubMed] [Google Scholar]834. Mendez MA, Garcia-Esteban R, Guxens M, Vrijheid M, Kogevinas M, Goñi F, Fochs S, Sunyer J. 2011. Prenatal organochlorine compound exposure, rapid weight gain, and overweight in infancy. Environ Health Perspect 119:272–278 [PMC free article] [PubMed] [Google Scholar]835. Cho MR, Shin JY, Hwang JH, Jacobs DR, Jr, Kim SY, Lee DH. 2011. Associations of fat mass and lean mass with bone mineral density differ by levels of persistent organic pollutants: National Health and Nutrition Examination Survey 1999–2004. Chemosphere 82:1268–1276 [PubMed] [Google Scholar]836. Monica Lind P, Lind L. 10 May 2011. Circulating levels of bisphenol A and phthalates are related to carotid atherosclerosis in the elderly. Atherosclerosis 10.1016/j.atherosclerosis.2011.1005.1001 [PubMed] [Google Scholar]837. Melzer D, Rice N, Depledge MH, Henley WE, Galloway TS. 2010. Association between serum perfluorooctanoic acid (PFOA) and thyroid disease in the U.S. National Health and Nutrition Examination Survey. Environ Health Perspect 118:686–692 [PMC free article] [PubMed] [Google Scholar]838. Trabert B, De Roos AJ, Schwartz SM, Peters U, Scholes D, Barr DB, Holt VL. 2010. Non-dioxin-like polychlorinated biphenyls and risk of endometriosis. Environ Health Perspect 118:1280–1285 [PMC free article] [PubMed] [Google Scholar]839. Kim KY, Kim DS, Lee SK, Lee IK, Kang JH, Chang YS, Jacobs DR, Steffes M, Lee DH. 2010. Association of low-dose exposure to persistent organic pollutants with global DNA hypomethylation in healthy Koreans. Environ Health Perspect 118:370–374 [PMC free article] [PubMed] [Google Scholar]840. Laclaustra M, Navas-Acien A, Stranges S, Ordovas JM, Guallar E. 2009. Serum selenium concentrations and diabetes in U.S. adults: National Health and Nutrition Examination Survey (NHANES) 2003–2004. Atherosclerosis 117:1409–1413 [PMC free article] [PubMed] [Google Scholar]841. Laclaustra M, Stranges S, Navas-Acien A, Ordovas JM, Guallar E. 2010. Serum selenium and serum lipids in US adults: National Health and Nutrition Examination Survey (NHANES) 2003–2004. Atherosclerosis 210:643–648 [PMC free article] [PubMed] [Google Scholar]842. Ahmed S, Mahabbat-e Khoda S, Rekha RS, Gardner RM, Ameer SS, Moore S, Ekström EC, Vahter M, Raqib R. 2011. Arsenic-associated oxidative stress, inflammation, and immune disruption in human placenta and cord blood. Environ Health Perspect 119:258–264 [PMC free article] [PubMed] [Google Scholar]843. Claus Henn B, Ettinger AS, Schwartz J, Téllez-Rojo MM, Lamadrid-Figueroa H, Hernández-Avila M, Schnaas L, Amarasiriwardena C, Bellinger DC, Hu H, Wright RO. 2010. Early postnatal blood manganese levels and children’s neurodevelopment. Epidemiology 21:433–439 [PMC free article] [PubMed] [Google Scholar]844. Wirth JJ, Rossano MG, Daly DC, Paneth N, Puscheck E, Potter RC, Diamond MP. 2007. Ambient manganese exposure is negatively associated with human sperm motility and concentration. Epidemiology 18:270–273 [PubMed] [Google Scholar]845. Lee DH, Lee IK, Porta M, Steffes M, Jacobs DR., Jr 2007. Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non-diabetic adults: results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia 50:1841–1851 [PubMed] [Google Scholar]
Tradução livre, parcial, de Luiz Jacques Saldanha, junho de 2020.


{kind=link}
{kind=link}